Алгоритм Ipex-2D для извлечения информации о компонентах пробы из массивов данных (ВЭЖХ-МС)-экспериментов протеомики
-экспериментов протеомики")
Автор: Макаров В.В., Лютвинский Я.И., Веренчиков А.Н.
Журнал: Научное приборостроение @nauchnoe-priborostroenie
Рубрика: Масс-спектрометрия для биотехнологии. Интерпретация данных, методология, применение
Статья в выпуске: 3 т.16, 2006 года.
Бесплатный доступ
Описан алгоритм обработки хромато-масс-спектрометрических данных с целью извлечения информации о молекулярных массах, времени хроматографического элюирования и интенсивности сигналов компонентов пробы. Алгоритм адаптирован для обработки масс-спектров многокомпонентных смесей пептидов, характерных для экспериментов протеомики. Описан исследовательский комплекс программ, в котором реализован данный алгоритм. Приводятся результаты тестирования алгоритма на масс-спектрометрических данных, полученных в модельном эксперименте по идентификации белка.
Короткий адрес: https://sciup.org/14264442
IDR: 14264442 | УДК: 621.384.668.8:
Ipex-2D: data mining algorithm for mass spectra of LC-MS experiment
An algorithm is described for extraction of analytically significant information from spectra of LC-MS experiment. The algorithm is adjusted for mass spectra of complex peptide mixtures, which are typical for proteomics experiments. The algorithm has been implemented in the research-grade software and tested in the example protein identification.
Текст научной статьи Алгоритм Ipex-2D для извлечения информации о компонентах пробы из массивов данных (ВЭЖХ-МС)-экспериментов протеомики
T t ' c 'n c e [ i ]
I е + I e • [ i ]
t
' e [ i ]
где индекс n обозначает обновленные значения параметров кластера.
В случае, если условие (1) не выполняется ни для одного из существующих кластеров, результат C [ i ] выделяется в отдельный кластер.
По завершении обработки кластеры тестируют-
. I ..
- _ _ -Л. _ * _ е [ j ] е [ j ]
■ *
. X '.[j]
e [ j ] e e [ i ]
У t. I.
. e [ j ] e [ j ]
_ e [ J ] e e [ i ]
. X - ex,]
e [ j ] e e [ i ]
.
,
Суммарная интенсивность сигнала ионов компонента пробы определяется по элементу кластера с максимальной интенсивностью:
C[z] .max ( 1 с'г.т) •
[ ] e [ , ] e e [ i ] e I J ]
РЕАЛИЗАЦИЯ АЛГОРИТМА
Для реализации и тестирования алгоритма IPEX-2D и других алгоритмов обработки масс-спектрометрических данных был разработан исследовательский комплекс программ "Масс-процессор". Создание данного комплекса программ позволило унифицировать форматы хранения и процедуры доступа к хромато-масс-спектрометрическим данным, а также реализовать инструменты их визуализации.
В состав комплекса (рис. 2) входят следующие функциональные блоки.
-
1. Реляционная база данных формата Microsoft SQL Server для хранения хромато-масс-спектрометрических данных и результатов их обработки.
-
2. Программа "Масс-процессор", в которой реализован графический интерфейс пользователя, средства доступа к данным, инструменты обработки данных, отображения и сохранения результатов.
-
3. Библиотеки методов обработки хромато-
- масс-спектрометрических данных.
-
4. Набор вспомогательных модулей для импортирования данных, полученных на оборудовании различных производителей.
Модульная структура графического интерфейса позволяет напрямую связать алгоритмы обработки данных с инструментами визуализации, обеспечить гибкость и расширяемость комплекса программ. Графический интерфейс программы "Масс-процессор" представлен на рис. 3. Доступ к базе экспериментальных данных организован в окне 1 в виде структуры. Визуализация масс-спектра осуществляется на одномерной диаграмме 2, в окне которой реализован стандартный набор инструментов масштабирования и выбора отображаемого диапазона шкалы. Для визуализации массива хромато-масс-спектрометрических данных разработан инструмент двумерной растровой диаграммы 3, которая строится в осях "Отношение массы иона к заряду—Время хроматографического элюирования", где логарифм интенсивности сигнала кодируется оттенками серого цвета различной плотности.
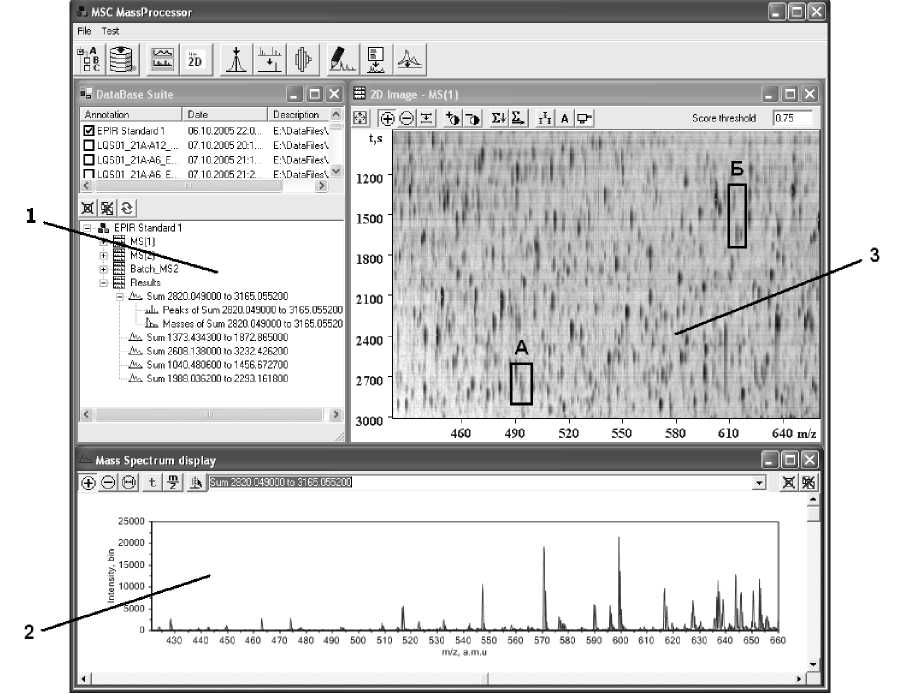
Рис. 3. Графический интерфейс программы "Масс-процессор". Пояснение в тексте
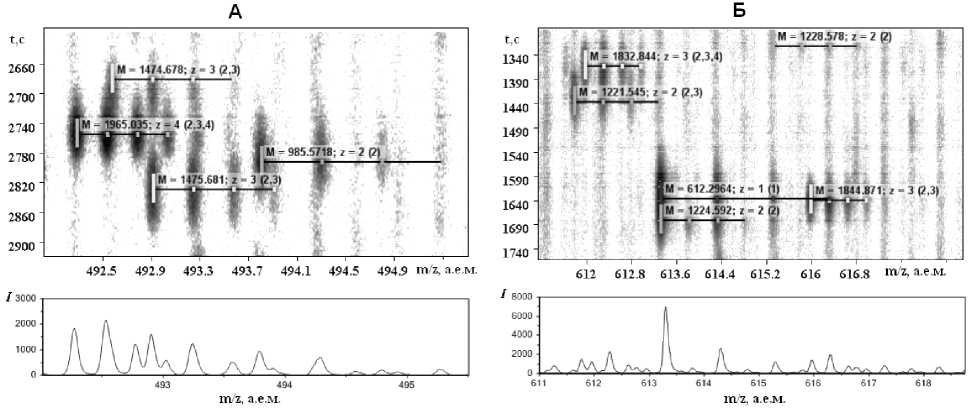
Рис. 4. Фрагменты массива хромато-масс-спектрометрических данных, представленного на рис. 3, и соответствующие суммарные масс-спектры
В графическом интерфейсе программы "Масс-процессор" на рис. 3 представлен массив хроматомасс-спектрометрических данных гидролизата модельной смеси белков (bovine serum albumin, bovin thyroglobulin, chicken ovalbumin, horse ferritin, fructose-bisphosfate aldolase, l-lactate dehydrogenase). На рис. 4 приведены результаты обработки данных алгоритмом IPEX-2D, иллюстрирующие способность алгоритма к выделению информации о компонентах пробы, сигналы которых претерпевают наложения в силу близких значений времени хроматографического элюирования и отношения массы к заряду. При визуальном анализе масс-спектров правильное выделение компонентов с молекулярными массами 1474.678, 1475.681, 1832.844, 1221.545 а.е.м. представляет значительные трудности по причине наложения спектральных пиков. При использовании алгоритма IPEX-2D данные компоненты были успешно выделены. Для каждого из перечисленных компонентов были обнаружены изотопные мультиплеты ионов нескольких зарядовых состояний, что свидетельствует о высокой достоверности выделения.
ИСПОЛЬЗОВАНИЕ АЛГОРИТМА IPEX-2D В ЭКСПЕРИМЕНТЕ ПО ИДЕНТИФИКАЦИИ БЕЛКА
Для тестирования алгоритма IPEX-2D был поставлен модельный эксперимент по идентификации белка методом пептидного массового картирования. В качестве модельного белка был выбран бычий сывороточный альбумин (БСА), широко используемый для тестирования аналитических характеристик масс-спектрометрических приборов.
Препарат БСА (Sigma, США) был гидролизо-ван трипсином, затем полученная смесь пептидов анализировалась методом ВЭЖХ-МС. Масс-спектрометрический анализ проводился на времяпро-летном масс-спектрометре МХ-5303 с источником ионов "электроспрей" (разработка Института аналитического приборостроения РАН). Предварительное разделение пробы производилось на жидкостном хроматографе "Милихром А-02" (производства ЗАО Институт хроматографии "Эконова", г. Новосибирск), работающем в режиме прямой стыковки с источником ионов.
В результате масс-спектрометрического эксперимента был получен массив из 569 масс-спектров, регистрация которых производилась в течение 21 мин с периодом около 2 с.
Массив экспериментальных данных был обработан алгоритмом IPEX-2D. Список из 367 компонентов пробы, полученный в результате обработки, был направлен в программу Mascot, доступную в сети Интернет по адресу для интерпретации по методу пептидного массового картирования. Проба была верно интерпретирована как гидролизат БСА. Данный вариант интерпретации оценен наивысшим значением рейтинга, в то время как оценки прочих гипотез лежат ниже порога достоверности, вычисляемого программой Mascot.
Пептиды, обнаруженные в пробе, в совокупности покрывают 71 % аминокислотной последова- тельности белка, что является высоким показателем для метода пептидного массового картирования. Высокая достоверность идентификации белка свидетельствует о качественном решении задачи извлечения аналитически значимой информации из масс-спектрометрических данных.
ЗАКЛЮЧЕНИЕ
Представленный алгоритм позволяет автоматизировать обработку хромато-масс-спектрометрических данных экспериментов протеомики. Высокая производительность алгоритма позволяет использовать его в программном обеспечении масс-спектрометрических приборов, разрабатываемых в настоящее время. Способность алгоритма к выделению информации о компонентах пробы, сигналы которых претерпевают наложения, позволяет успешно анализировать смеси с большим количеством компонентов, что особенно актуально для задач протеомики.
