Аналитический комплекс для определения аминокислотной последовательности пептидов

Автор: Назимов И.В., Краснов Н.В., Новиков А.В., Бубляев Ростислав Анатольевич, Фиронов С.В., Присяч С.С., Мурадымов М.З.
Журнал: Научное приборостроение @nauchnoe-priborostroenie
Рубрика: Приборы и измерительные методы для биохимии
Статья в выпуске: 4 т.21, 2011 года.
Бесплатный доступ
В работе продемонстрированы возможности отечественного аналитического комплекса, позволяющего хроматографически разделять смеси пептидов на индивидуальные компоненты, масс-спектрометрически фрагментировать последние и производить вероятностную идентификацию аминокислотной последовательности на основании протеомных баз данных. Возможности подхода продемонстрированы на примере анализов пептидов из гемоглобина человека и из ацетилхолинового рецептора человека.
Масс-спектрометрия, пептиды, аминокислотная последовательность
Короткий адрес: https://sciup.org/14264752
IDR: 14264752 | УДК: 621.384.668.8:
Analytical station for peptide amino acid sequencing
The paper describes possibilities of the domestic analytical station allowing to divide a mixture of peptides by HPLC on individual components, followed by mass spectrometric fragmentation of the separated peptides and to make stochastic identification of amino acid sequences basing on the proteome databases comparison. Station possibilities are demonstrated on the peptide analyses of human hemoglobin and human acetylcholine receptor fragments.
Текст научной статьи Аналитический комплекс для определения аминокислотной последовательности пептидов
В настоящее время для определения аминокислотной последовательности пептида почти во всех случаях используют масс-спектр его фрагментов (обычно для этого используют MALDI-MS-MS приборы). Аминокислотная последовательность подбирается на основании разницы масс между ионами фрагментов исходного (родительского) пептида. В идеальном варианте масс-спектрометр регистрирует весь набор масс, полученный последовательным отщеплением одного за другим аминокислотных остатков от родительского иона с обоих концов. Фрагментные ионы с сохраненным С-концом пептида принято называть Y-ионами, с сохраненным N-концом пептида — B-ионами (рис. 1).
Такая методология позволяет определить последовательность de novo до 8 аминокислот с достоверностью 99 %. Отличительными особенностями такого подхода являются относительная простота спектра фрагментных ионов и приемлемая надежность интерпретации спектров [1]. Однако большая зашумленность MALDI-масс-спектров в области малых масс ионов (до 300– 400 Да) существенно затрудняет проведение анализа ди- и трипептидов. Также следует отметить сложность и высокую стоимость тандемного масс-спектрометра.
Кроме того, решение задачи нахождения последовательности пептида из восьми аминокислотных остатков по его фрагментному масс-спектру "в лоб", полным перебором всех вариантов последовательности потребует как минимум 5∙1010 операций (20 вариантов аминокислотных остатков в каждой из восьми позиций последовательности). Последующее же увеличение длины
|
В1 |
в2 |
Вз |
B4 |
B5 |
|
нн2 1 |
nh2 1 — |
nh2 ____-1_____ |
nh2 1 — |
nh2 |
|
G |
G |
G |
G |
1 ® |
|
СО* |
CO |
CO |
CO |
co |
|
58.0 |
L |
L |
L |
L |
|
CO+ |
CO |
CO |
co |
|
|
171.1 |
s |
s |
s |
|
|
CO+ |
co |
co |
||
|
МН3+ |
258.1 |
D |
D |
|
|
L |
446.3 |
CO* 373.2 |
CO |
|
|
СО |
NH3+ |
G |
||
|
5 |
CO+ |
|||
|
359 3 |
430.2 |
|||
|
со |
co |
NH3+ |
||
|
D |
D |
I) |
244.2 |
|
|
СО |
CO |
CO |
NHyb |
|
|
G |
G |
G |
G |
187 2 |
|
СО |
CO |
CO |
CO |
NH3+ |
|
W |
w |
w |
w |
W |
|
сдои |
COOH |
COOH |
COOH |
COOH |
|
Ye |
y4 |
Y3 |
Yi |
Yi |
Рис. 1. Серия фрагментных B- и Y-ионов, образованная отщеплением по одному аминокислотному остатку от родительского иона GLSDGW последовательности на 1 аминокислотный остаток будет увеличивать вычислительную нагрузку более чем на порядок (минимум в 20 раз).
Среди подходов, позволяющих снизить вычислительную нагрузку на ЭВМ, в настоящее время наибольшее признание получило прямое вероятностное сравнение фрагментных масс-спектров с аминокислотными последовательностями из баз данных. Такой подход дает преимущество непосредственной идентификации пептида. Однако накладывает и ограничения, делая невозможным определение последовательности de novo . Существенно нивелировать этот недостаток при сравнении фрагментных масс-спектров позволяет использование открытых (не связанных лицензионными ограничениями, бесплатных) протеомных (белковых) баз данных. Рано или поздно в такие базы данных на основе анализа генетического кода попадают все пептиды с известной или предполагаемой аминокислотной последовательностью. Таким образом, круг сравнения расширяется до всех известных пептидов.
СОСТАВ И ВОЗМОЖНОСТИ АНАЛИТИЧЕСКОГО КОМПЛЕКСА
Целью данной работы являлась апробация отечественного аналитического комплекса вероятностной идентификации пептидов на основании их фрагментных масс-спектров. Комплекс был разработан в Институте аналитического приборостроения РАН по целевой научно-технической программе "Разработка уникальных научно-исследовательских приборов и оборудования для учреждений РАН" в 2006–2010 гг. В состав аналитического комплекса вошли высокоэффективный жидкостный микроколоночный хроматограф "Мили-хром А-02", настольный времяпролетный масс-спектрометр МХ5311 с источником ионов электроспрей с ортогональным вводом ионов (ВЭЖХ-ВПМС) и программно-аппаратный комплекс "Proteos" для распознавания аминокислотных последовательностей пептидов по их фрагментным масс-спектрам.
Микроколоночный жидкостной хроматограф "Милихром А-02" использовался в режиме прямой стыковки ( on line) с времяпролетным масс-спектрометром МХ5311. Электрогазодинамическая система транспортировки источника ионов "электроспрей" позволяет управлять степенью фрагментации молекулярного иона при помощи изменения электрического поля.
Экспериментально определено, что для получения информативного масс-спектра необходимо снять 5–6 масс-спектров фрагментных ионов пептида при разных значениях электрического поля в газодинамической системе транспортировки источника ионов для получения различной степени фрагментации, что является достаточным для однозначной интерпретации масс-спектра фрагментов пептида и последующего определения его полной аминокислотной последовательности. Отмеченные закономерности подтверждены результатами анализа аминокислотной последовательности нескольких десятков пептидов различной длины и аминокислотного состава, что позволяет сделать вывод о принципиальной возможности идентификации аминокислотной последовательности пептидов, содержащих до 10–15 аминокислотных остатков, что соответствует средней длине пептидов — продуктов ферментативного гидролиза белков, которые являются объектами как фундаментальных протеомных исследований, так и прикладных генно-инженерных процессов. Экспериментальные результаты позволяют надеяться на то, что данный метод определения аминокислотной последовательности по своей экономичности, чувствительности анализа, производительности, информативности является конкурентоспособным по сравнению с другими вариантами определения аминокислотной последовательности пептидов.
Вычислительный комплекс представляет собой отдельный компьютер серверного типа со специальным программным обеспечением, основанным на технологиях распознавания и поиска PST (Peptide Sequence Tag) [2, 3], разрабатываемых ИАП РАН. Вычислительный комплекс производит идентификацию пептидов по их масс-спектрам в формате представления PKL, полученным на следующих типах приборов: Q-TOF; IT-FT-ICR; MALDI-TOF-TOF; ESI-o-TOF (МХ5311), с реализацией режима фрагментации коротких пептидов. От приборов требуются рядовые по нынешним меркам технические характеристики: разрешающая способность не хуже 3000; массовая точность не хуже 50 ppm; динамический диапазон — не менее 300. Вычислительный комплекс производит поиск, в том числе error-tolerant (устойчивый к выпадению, добавлению и модификации аминокислотных остатков в последовательности пептида), на основании распознанных PST в протеом-ных базах данных Swiss-prot, Trembl, NCBI NRDB, NCBI dbEST. Комплекс производит идентификацию пептидов, несущих пост-трансляционные модификации, в том числе определенные пользователем, по их масс-спектрам.
Результатом работы вычислительного комплекса является формирование гипотез о возможных исходных последовательностях аминокислот, входящих в состав исходной смеси пептидов, на основании сравнения фрагментного масс-спектра и открытых протеомных баз данных.
ПРИБОРЫ, МАТЕРИАЛЫ И МЕТОДЫ
Для растворения образцов и хроматографического анализа использовали ацетонитрил ("Криохром", Санкт-Петербург, сорт 1 или 2), муравьиную кислоту ("Merck", ФРГ). Тридистиллят воды, используемый для приготовления растворов, и ацетонитрил пропускали через фильтры 0.45 мкм "Millipore" (США).
Аналитические параметры настольного ВПМС МХ5311 во время эксперимента составляли по разрешающей способности не менее 5500, по массовому диапазону до 10 000 Да, массовая точность 10 ppm, стабильность определения массы в течении часа не хуже 10 ppm при скорости сканирования 50 спек./с.
Изучаемые образцы растворов пептидов хроматографировали на колонке с обращенной фазой (Prontosil C18: 120 мкм, 2.0 × 75 мм, Институт хроматографии "Эконова", Новосибирск) в градиенте концентрации ацетонитрила. Элюат, содержащий компоненты смеси, через металлический капилляр диаметром 100 мкм, подавался в источник ионов — электроспрей со скоростью 2– 50 мкл/мин.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Пептиды гемоглобина человека получали по следующей методике. Цельную кровь человека промывали физиологическим раствором (5 раз при комнатной температуре). Отмытый осадок расфасовывали по порциям 5 мл, хранили при –20 °С, размораживая порции осадка по мере необходимости для работы.
Размороженную порцию заливали 0.2 М уксусной кислотой, оставляли при комнатной температуре на 4–6 часов (время гидролиза гемоглобина определялось потребностями получения числа и длины (длинных или коротких) пептидов гидролизата). Полученный гидролизат центрифугировали при комнатной температуре (10000 об./мин, 10 мин). Осадок отбрасывали, надосадочную жидкость (раствор смеси пептидов) использовали для разделения компонентов и выделения нужных индивидуальных пептидов для масс-спектрометрического анализа.
Получившийся гидролизат гемоглобина (набор пептидов) подвергали хроматографическому разделению, для чего использовали 0.2 % раствор муравьиной кислоты с градиентом концентрации ацетонитрила от 0 до 100 % и детекции компонентов при 206 и 280 нм (рис. 2). Фракции разделенных пептидов в том же элюате вводились в масс-спектрометр и подвергались фрагментации в источнике ионов воздействием регулируемого электрического поля. Критерием оптимальности выбранного режима фрагментации являлось равномерность заполнения масс-спектра пиками фрагментов от родительского иона до фрагмент-ных ионов с массами в области 150–200 Да.
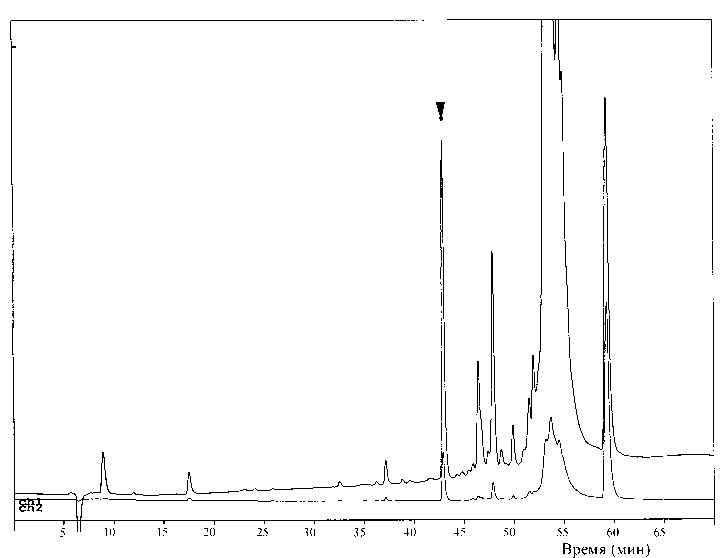
Рис. 2. Разделение пептидов лизата гемоглобина крови человека.
Стрелкой отмечен N-концевой пептид альфа-цепи гемоглобина человека (аминокислотные остатки 1–33). Кривые: ch1 — детекция на 206 нм, ch2 — 280 нм
Формирование гипотез о возможных последо- теомных баз данных (таких как NRDB и Swiss-вательностях аминокислот на основании сравне- prot) проводили при помощи комплекса Proteos. ния фрагментного масс-спектра и открытых про-
|
Tag |
Est. |
Exact |
Inexact |
|
18 [766.385XAGYE[2158.01] |
0.0247278 |
||
|
19 [4l2.207]PTREK[2450.13] |
0.0242145 |
||
|
20 [2150.04]NRNKT[710.262] |
0.0210566 |
||
|
21 [380.189]ETREK[2450.13] |
0.0206721 |
||
|
22 [2264.07]NRKTE[581.202] |
0.0204481 |
||
|
23 [2662.29]TENAP[299.111] |
0.0200133 |
Search tags! |
|
Peptide |
Est. |
||
|
2 |
VL5PADQTNVQAAWGQVGAHAGEYGAEALERMF |
0.999509 |
>gi I P69906|tid|9597|nm| Hemoglobin subunit alpha (Hemoglobin |
|
3 |
VVLAY5GGLDT51LLQWIQTEYGAEVLTFTAD |
0.998323 |
>gi|Q9A6Ul |tid| l55892|nm|Argininosuccinate synthase (EC 6.3 |
|
4 |
VL5PADQTIWQTAWGQVGGHGGEYGAEALERMF |
0.979716 |
>gi|P01930|tid|164648|nm|Hemoglobin subunit alpha (Hemoglob |
|
5 |
L5ADDQANLQATWEQLGGHGAEYGAEALERMF |
0.950082 |
>gi|P19014|tid|10085|nm|Hemoglobin subunit alpha (Hemoglobir |
|
6 |
LSAADQNNVQGLFTQLAGHAEEYGAEALERMF |
0.92956 |
>gi|P01995|tid|9057|nm|Hemoglobin subunit alpha-A (Hemoglob |
|
< |
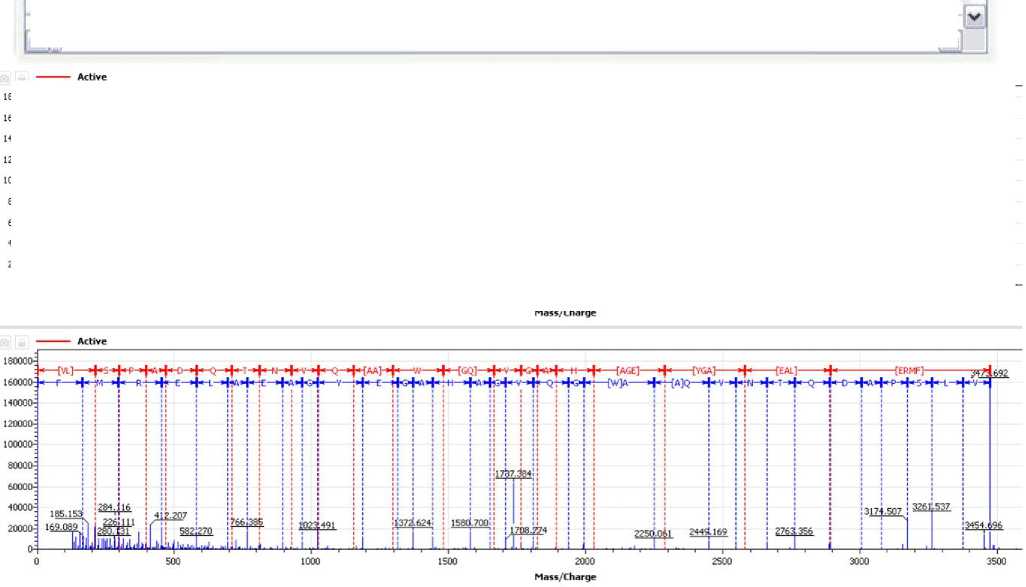
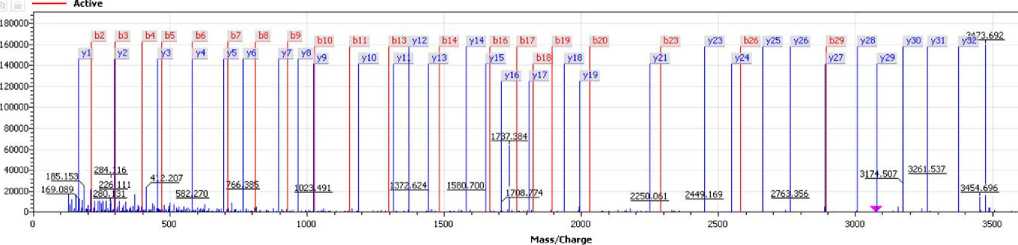
Рис. 3. Найденные теги (вероятные части искомой аминокислотной последовательности), выявленные на фрагментном масс-спектре N-концевого пептида альфа-субъединицы гемоглобина человека
ОБСУЖДЕНИЕ РЕЗУЛЬТАТОВ
Получение гомогенных фракций пептидов в ходе разделения смесей пептидов может значительно ускорить определение их аминокислотной последовательности при одновременном увеличении степени достоверности ее определения.
С точки зрения эффективности разделения вышеуказанные условия (6000–7000 теоретических тарелок на колонку диаметром 2.0 мм и длиной 75–150 мм) достаточны для полного разделения смесей, содержащих 25–30 пептидов, на индивидуальные компоненты. Реально серийные колонки фирмы "Эконова" (2 × 75 мм) имеют 5000– 6000 т.т., что при оптимизации профиля элюирования позволяет выделить на них в гомогенном виде 80–90 % пептидов из смеси упомянутой сложности. Такой эффективности хроматографического разделения достаточно для успешного решения большинства задач масс-спектрометрического структурного анализа пептидов даже в случае минимальной пробоподготовки биологического материала.
Например, в результате масс-спектрометрического фрагментационного анализа полипептида из ацетилхолинового рецептора человека его аминокислотная последовательность подтверждена с высокой степенью достоверности (99 %).
Несмотря на достаточно высокую сложность компонентного состава лизата гемоглобина крови человека на упомянутой колонке удалось хроматографически выделить в чистом виде, а после масс-спектрометрического анализа с помощью программного комплекса "Proteos" идентифицировать N-концевой пептид альфа-цепи гемоглобина человека (33 аминокислотных остатка, рис. 3).
ВЫВОДЫ
Использование отечественного аналитического комплекса, включающего микроколоночный жидкостный хроматограф "Милихром А-02", время-пролетный масс-спектрометр с источником ионов "электроспрей" МХ5311, и аппаратно-программного комплекса "Proteos" позволяет разделить пептидные компоненты сложных смесей, определить величину их молекулярной массы, управляемо фрагментировать исходную молекулу и, прове- дя отнесение с базами данных, распознавать аминокислотную последовательность пептида.
На описанный метод подана патентная заявка № 2011137344 от 31.08.2011:
Назимов И.В., Краснов Н.В., Новиков А.В., Бубля-ев Р.А., Фиронов С.В., Присяч С.С., Мурадымов М.З. "Метод масс-спектрометрического секвенирования пептидов и определение их аминокислотных последовательностей".
В настоящее время к программно-аппаратному комплексу "Proteos" предоставляется свободный доступ в Интернете по адресу URL: (http://195.182.140.214/proteos).
