Анализ генетического разнообразия, структуры и дифференциации популяций Pinus sylvestris L. на Урале

Автор: Чертов Н.В.
Журнал: Вестник Пермского университета. Серия: Биология @vestnik-psu-bio
Рубрика: Генетика
Статья в выпуске: 2, 2024 года.
Бесплатный доступ
Проведен молекулярно-генетический анализ девяти популяций Pinus sylvestris L. на Урале с использованием ISSR (Inter Simple Sequence Repeats) анализа полиморфизма ДНК. Установлен 141 фрагмент ДНК, из которых 8 (5.67%) являются уникальными. У изученных популяций сосны обыкновенной определен средний уровень генетического разнообразия (P95 = 0.886; I = 0.224; HE = 0.141; ne = 1.301). Выявлено, что две популяции из Южного Урала характеризуются сниженным генетическим разнообразием, это популяции из Салаватского (P95 = 0.746; I = 0.142; HE = 0.091; ne = 1.153) и Мечетлинского (P95 = 0.702; I = 0.117; HE = 0.071; ne = 1.106) р-нов Республики Башкортостан. Анализ генетической структуры показал, что изученные популяции подразделяются на 4 кластера, в целом соответствующие их географическому расположению. Установлено, что изученные популяции характеризуются средней степенью дифференциации (GST = 0.315). Корреляционный анализ выявил среднюю положительную корреляцию между географическими и генетическими расстояниями (R2 = 0.358). Результаты исследования могут быть использованы для составления программ по восстановлению и эффективному использованию лесных генетических ресурсов на Урале.
Полиморфизм днк, генетическое разнообразие, генетическая структура, pinus sylvestris l, урал
Короткий адрес: https://sciup.org/147244925
IDR: 147244925 | УДК: 575.174.015.3 | DOI: 10.17072/1994-9952-2024-2-221-230
Analysis of genetic diversity, structure and differentiation of Pinus sylvestris L. populations in the Urals
The article is devoted to a molecular genetic analysis of nine populations of Pinus sylvestris L. in the Urals was carried out using ISSR (Inter Simple Sequence Repeats) DNA polymorphism analysis. 141 DNA fragments were identified, of which 8 (5.67%) are unique. The studied populations of Scots pine revealed an average level of genetic diversity (P95 = 0.886; I = 0.224; HE = 0.141; ne = 1.301). It was revealed that two populations from the southern part of the Southern Urals are characterized by reduced genetic diversity, these are populations from Salavat (P95 = 0.746; I = 0.142; HE = 0.091; ne = 1.153) and Mechetlinsky (P95 = 0.702; I = 0.117; HE = 0.071; ne = 1.106) districts of the Republic of Bashkortostan. Analysis of the genetic structure showed that the studied populations are divided into 4 clusters, generally corresponding to their geographical location. It was found that the studied populations are characterized by an average degree of differentiation (GST = 0.315). Correlation analysis revealed an average positive correlation between geographic and genetic distances (R2 = 0.358). The results of the study can be used to draw up programs for the restoration and efficient use of forest genetic resources.
Текст научной статьи Анализ генетического разнообразия, структуры и дифференциации популяций Pinus sylvestris L. на Урале
Генетическое разнообразие важно для выживания видов и играет решающую роль в их сохранении [Spielman, Brook, Frankham, 2004; O'Grady et al., 2006; Frankham, 2012]. Для поддержания адаптивного потенциала необходимо сохранять аллельное разнообразие популяций, включая как типичные, так и специфичные аллели [Потокина, Александрова, 2008].
Хвойные леса составляют основу бореальных экосистем и имеют огромное экономическое значение. Они оказывают локальное и глобальное влияние на экосистемы, являясь важной частью регулирования водного стока и сохранения почв, важнейшим звеном в круговороте углерода и средством очистки атмосферного воздуха от загрязнений [Högberg et al., 2001; Pan et al., 2011; Lindén et al., 2014]. Кроме этого, ткани хвойных растений богаты биологически активными веществами (BAC, Biologically Active Compounds), такими как терпеноиды, стероиды, алкалоиды, флавоноиды, комплексы полисахаридов (холоцеллюлоза) и прочее, являющиеся перспективным сырьем для фармацевтической промышленности [Dering et al., 2017; Liu et al., 2019].
Сосна обыкновенная ( Pinus sylvestris L.) – вторая по распространенности хвойная порода в мире, имеющая большое экономическое и экологическое значение. Сосновые леса широко распространены в мире и, в частности, в Северном полушарии, что делает сосну обыкновенную одной из важнейших лесообразующих пород [Floran, Sestras, Gil, 2011]. Современный ареал этого вида является результатом событий повторной колонизации и послеледникового сокращения некогда большего ареала распространения [Hebda, Wójkiewicz, Wachowiak, 2017; Toth et al., 2017]. P. sylvestris — вид, толерантный к широкому спектру экологических условий окружающей среды и играющий важную экономическую и экологическую роль в лесных экосистемах Европы [Hogberg et al., 2001; Lindén et al., 2014]. Молекулярногенетические исследования P. sylvestris проводились как в Европе, так и в России [Floran, Sestras, Gil, 2011; Видякин и др., 2012; Санников, Петрова, 2012; Pazouki et al., 2016; Hebda, Wójkiewicz, Wachowiak, 2017; Toth et al., 2017; Chertov et al., 2022; Kavaliauskas, Danusevičius, Baliuckas, 2022]. С использованием аллозимного анализа были изучены полиморфизм и дифференциация популяций P. sylvestris, филогео-графическая структура, а также установлены плейстоценовые рефугиумы вида [Санников, Петрова, 2012]. Результаты анализа митохондриальных маркеров свидетельствуют о том, что на всем пространстве от востока Восточно-Европейской равнины и по крайней мере до реки Енисей, вид практически генетически однороден [Видякин и др., 2012]. На Северном и Среднем Урале молекулярно-генетические исследования P. sylvestris носят фрагментарный характер и велись в области молекулярно-генетической идентификации популяций [Боронникова и др., 2018; Пришнивская и др., 2019; Чертов и др., 2020].
Молекулярно-генетический анализ генетического разнообразия древесных видов растений с использованием ISSR (Inter Simple Sequence Repeats) является простым и доступным ПЦР-методом анализа полиморфизма ДНК [Zietkiewicz, Rafalski, Labuda, 1994]. Из-за большого числа копий микросателлитных последовательностей и их большого числа в геномах эукариот использование последовательностей SSR (Simple Sequence Repeats) в качестве праймера для метода фингерпринтинга ДНК на основе ПЦР удобно и эффективно [Kalendar et al., 2019; Kalendar, Muterko, Boronnikova, 2021].
Сосна обыкновенная широко используется в хозяйственной деятельности, ее древесина активно заготавливается. Для составления программ рационального использования лесных ресурсов необходимы знания о генетическом разнообразии и структуре популяций, произрастающих на участках активных рубок, полученные путем выявления полиморфных фрагментов ДНК. В связи с этим изучение молекулярно-генетического разнообразия и генетической структуры популяций P. sylvestris Урала с использованием ISSR-метода перспективно для разработки и оптимизации протоколов оценки состояния генофондов популяций P. sylvestris Урала. Таким образом, настоящая работа направлена на детальное изучение генетического разнообразия, генетической структуры и дифференциации природных популяций P. sylvestris в условиях их естественного произрастания в пределах Урала.
Материал и методика
В качестве объектов исследования были избраны 9 популяций сосны обыкновенной ( Pinus sylvestris L., Pinaceae). На Северном Урале и в Предуралье исследованы 4 популяции P. sylvestris, расположенные в Пермском крае: в Чердынском р-не ( PS_Ch ), в Гайнском р-не ( PS_Gn ), в Кочёвском р-не ( PS_Kh ), в Усольском р-не ( PS_Rm ). На Среднем Урале находятся 3 изученные популяции: в Кудымкарском р-не
( PS_Ln ) и в Кишертском р-не ( PS_Pr ), Пермского края, а также в Каслинском р-не Челябинской обл. ( PS_Ar ). На Южном Урале находятся 2 популяции: в Мечетлинском ( PS_Mh ) и в Салаватском р-нах ( PS_Sl ) Республики Башкортостан. Сбор растительного материала проводили с деревьев, расположенных на расстоянии не менее 50–150 м друг от друга. Географические расстояния между популяциями варьировали от минимальных 87 км между PS_Kh и PS_Gn до максимальных 628 км между популяциями PS_Gn и PS_Sl (рис. 1). Высота над уровнем моря варьировала от 117 ( PS_Rm ) до 338 м ( PS_Ar ).
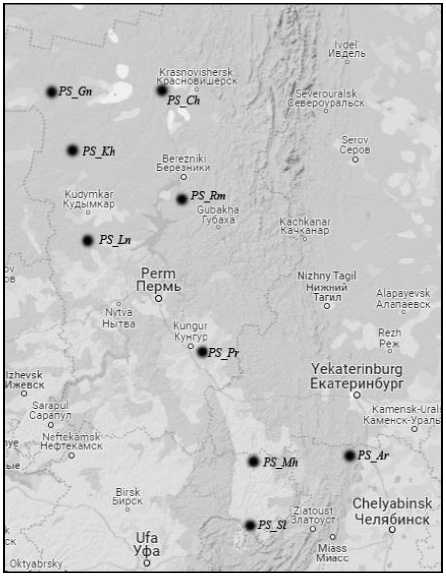
Рис. 1. Карта-схема расположения изученных популяций P. sylvestris :
PS_Ch – Чердынский р-н, PS_Gn – Гайнский р-н, PS_Kh – Кочёвский р-н, PS_Rm – Усольский р-н, PS_Ln – Кудымкарский р-н, PS_Pr – Кишертский р-н, PS_Ar – Каслинский р-н, PS_Mh – Мечетлинский р-н, PS_Sl – Салаватский р-н
[Schemаtic map of the location of the studied populations of P. sylvestris :
PS_Ch – Cherdynskyi district, PS_Gn – Gainskyi district, PS_Kh – Kochevskyi district, PS_Rm – Usolskyi district, PS_Ln – Kudymkarskyi district, PS_Pr – Kishertskyi district, PS_Ar – Kislinskyi district, PS_Mh – Mechetlinskyi district, PS_Sl – Salavatskyi district]
Образцы тканей растений для исследования были собраны индивидуально с 31 дерева в каждой из изучаемых популяций. Выделение ДНК проводили CTAB-методом [Rogers, Bendich, 1985]. Масса каждого образца сухого растительного материала составляла 20 мг. Для определения концентрации и качества ДНК использовали спектрофотометр NanoDrop™ 2000 (Thermo Fisher Scientific Inc., Waltham, MA, USA). Для полимеразной цепной реакции (ПЦР) пробы ДНК в каждом образце разбавляли до концентрации 10 нг/мкл.
Для оценки генетического разнообразия и генетической структуры популяций использовался ISSR-метод анализа полиморфизма ДНК. ПЦР проводили в реакционной смеси объемом 25 мкл, содержащей 25 нг матричной ДНК, 1 × ПЦР-буфер с 2.5 мМ MgCl2, 1 мкМ ISSR праймера, по 0.25 мМ каждого dNTP и 2 ед. ДНК-полимеразы Taq («Силекс М», Россия). Амплификацию проводили в термоциклере SimpliAmp™ (Thermo Fisher Scientific Inc., Waltham, MA, USA) при следующих условиях: начальная стадия денатурации при 94 °C в течение 2 минут, за которой следовали 32 цикла при 94 °C в течение 20 с, при 52–64 °С (в зависимости от последовательности праймера) в течение 30 с и при 72 °С в течение 60 секунд с последующей финальной элонгацией при 72 °С в течение 3 минут.
Продукты ПЦР разделяли электрофорезом при 120 В в течение 3 часов в 1.7% агарозном геле с буфером 1xTBE, окрашивали бромистым этидием и фотографировали в проходящем ультрафиолетовом свете с использованием системы документации геля Gel Doc XR+ (Bio-Rad Laboratories, Inc., Hercules, CA, USA). Для определения длины фрагментов ДНК использовали маркер молекулярной массы (100 п.н. + 1.5 + 3 Кб DNA Ladder, ООО. СибЭнзим-М, Москва, Россия) и программу Quantity One 1-D Analysis (Bio-Rad Laboratories, Inc.). Всего проанализирован полиморфизм 141 фрагмента ДНК с 5 праймерами у
278 отдельных деревьев P. sylvestris . Для проверки достоверности полученных результатов ПЦР проводили не менее трех раз.
Для количественной оценки генетического полиморфизма и определения генетической структуры девяти изученных популяций данные были представлены в виде матрицы бинарных данных, в которой учитывалось наличие или отсутствие в спектрах фрагментов одинакового размера как состояние 1 или 0 соответственно. Компьютерную обработку данных проводили с использованием специализированного макроса GenAlEx для MS Excel для определения абсолютного числа аллелей ( n a ), эффективного ( n e ) числа аллелей [Peakall, Smouse, 2006], ожидаемой ( H E ) гетерозиготности, информационного индекса Шеннона ( I ), доли полиморфных локусов ( P 95 ) и числа уникальных аллелей ( Un ). Для описания генетической структуры популяций [Nei, 1975]. использовали следующие параметры, рассчитанные в программе POPGENE 1.31 [Yeh et al., 1996]: ожидаемая доля гетерозиготных генотипов во всей популяции как мера общего генетического разнообразия ( H T ); ожидаемая доля гетерозиготных генотипов в субпопуляции как мера внутрипопуляционного разнообразия ( H S ); доля межпопуляционного генетического разнообразия в общем разнообразии или коэффициент генетической дифференциации ( G ST ); и AMOVA (Analysis of Molecular Variance) с расчетом индекса PhiPT (индекс подразделённости популяции) [Nei, Li, 1979]. Для определения корреляции между парными генетическими расстояниями (PhiPT) и географическими расстояниями в общей группе населения использовался тест Мантела.
На основе бинарной матрицы данных была рассчитана матрица генетических расстояний, на основе которой с помощью программы MEGA X была сгенерирована дендрограмма, отражающая степень сходства между изучаемыми популяциями [Kumar et al., 2018]. Кроме того, для проверки полученных данных был проведен анализ главных координат (PCoA), реализованный в программе PAST 4.10 [Hammer, Harper, Paul, 2001].
Результаты и их обсуждение
Молекулярно-генетический анализ девяти популяций P. sylvestris выявил 141 ПЦР-фрагмент, из них 125 были полиморфны. Используемые праймеры амплифицировали от 21 до 38 ПЦР-фрагментов, максимальное число фрагментов амплифицировалось с использованием праймера CR-212 [(CA) 6 GT]. В среднем один праймер идентифицировал 28 ПЦР-фрагментов. Длина ПЦР-фрагментов варьировала от 180 до 1500 пар оснований. Наибольшее генетическое разнообразие наблюдается в популяциях PS_Pr ( P 95 = 0.869; I = 0.289; H E = 0.184; n e = 1.294) из Кишертского р-на и PS_Ch ( P 95 = 0.911; I = 0.287; H E = 0.181; n e = 1.286) из Чердынского р-на. Наименьшее генетическое разнообразие отмечено в популяциях PS_Sl ( P 95 = 0.746; I = 0.142; H E = 0.091; n e = 1.153) из Салаватского р-на, PS_Mh ( P 95 = 0.702; I = 0.117; H E = 0.071; n e = 1.106) из Мечетлинского р-на Республики Башкортостан (табл. 1).
Таблица 1
Генетическое разнообразие изученных девяти популяций P. sylvestris
[Genetic diversity of the nine P. sylvestris populations studied]
|
Популяции |
P 95 |
H E |
n a |
n e |
I |
Un |
|
PS_Kh |
0.865 |
0.167 (0.014) |
1.716 (0.452) |
1.256 (0.290) |
0.271 (0.020) |
0 |
|
PS_Gn |
0.820 |
0.153 (0.014) |
1.681 (0.469) |
1.235 (0.292) |
0.247 (0.020) |
0 |
|
PS_Pr |
0.869 |
0.184 (0.015) |
1.695 (0.462) |
1.294 (0.323) |
0.289 (0.021) |
0 |
|
PS_Ln |
0.780 |
0.152 (0.014) |
1.667 (0.473) |
1.233 (0.289) |
0.246 (0.020) |
0 |
|
PS_Ch |
0.911 |
0.181 (0.015) |
1.695 (0.462) |
1.286 (0.312) |
0.287 (0.021) |
0 |
|
PS_Rm |
0.784 |
0.160 (0.014) |
1.660 (0.475) |
1.249 (0.300) |
0.256 (0.020) |
0 |
|
PS_Sl |
0.746 |
0.091 (0.014) |
1.376 (0.486) |
1.153 (0.302) |
0.142 (0.020) |
1 |
|
PS_Mh |
0.702 |
0.071 (0.011) |
1.362 (0.482) |
1.106 (0.217) |
0.117 (0.017) |
2 |
|
PS_Ar |
0.754 |
0.108 (0.015) |
1.312 (0.465) |
1.183 (0.318) |
0.162 (0.022) |
5 |
|
На общую выборку |
0.886 |
0.141 (0.005) |
2.000 |
1.301 (0.260) |
0.224 (0.007) |
8 |
Примечания: H E – ожидаемая гетерозиготность; n a – абсолютное число аллелей на локус; n e – эффективное число аллелей на локус; I – информационный индекс Шеннона; Un – число уникальных аллелей; у всех вышеуказанных параметров в скобках даны стандартные отклонения.
Общими для всех изученных популяций P. sylvestris из 141 установленных фрагментов ДНК являются 133 (94.33%). Только 8 фрагментов ДНК (5.67%) были уникальными ( Un ), т.е. встречающиеся только в одной популяции. Уникальные аллели были обнаружены в популяциях из Салаватского, Мечетлинского и Каслинского р-нов, 1, 2 и 5 аллелей соответственно (табл. 1).
При исследовании четырех популяций сосны обыкновенной на Южном Урале было выявлено 96 фрагментов ДНК, из них 79 были полиморфны [Khanova et al., 2020]. Это число фрагментов ДНК меньше, чем в изученных нами популяциях на Урале. Это может быть обусловлено тем, что изучено меньшее число выборок на Южном Урале. Тем не менее, по таким параметрам как ожидаемая гетерозиготность ( H E = 0.239) и эффективное число аллелей ( n e = 1.385) популяции Южного Урала более генетически разнообразны [Khanova et al., 2020].
В целом изученные популяции характеризуются средними показателями генетического разнообразия ( P 95 = 0.886; I = 0.224; H E = 0.141; n e = 1.301). Данный уровень генетического разнообразия ниже, чем у популяций Южного Урала ( H E = 0.239), Оренбургского Зауралья ( H E = 0.161) и некоторых популяций Восточно-Европейской равнины ( H E = 0.170) [Рябухина и др., 2019; Khanova et al., 2020; Сбоева, 2023]. Подобные различия могут быть связаны со степенью антропогенной нагрузки, в особенности интенсивностью лесозаготовок.
Анализ генетической структуры изученных популяций P. sylvestris показал, что ожидаемая доля гетерозиготных генотипов ( H T ) в общей выборке составила 0.205, тогда как ожидаемая доля гетерозиготных генотипов в субпопуляции ( H S ) меньше и составила 0.141. Коэффициент подразделения популяции ( G ST ) показывает, что на межпопуляционную составляющую приходится 0.315 от общего генетического разнообразия (табл. 2). Значения парных генетических расстояний PhiPT, обнаруженных пакетом AMOVA, находились в диапазоне от 0.080 ( PS_Kh/PS_Pr ) до 0.570 ( PS_Sl/PS_Ar ). Для всей выборки P. sylvestris индекс PhiPT составил 0.371, что близко к G ST = 0.315. Анализ молекулярной изменчивости (AMOVA) показал, что на межпопуляционную компоненту приходится 37% всего разнообразия, а на внутрипопуляционную – 63%. Данный уровень дифференциации ниже, чем у популяций Восточно-Европейской равнины [Видякин и др., 2015]. Это может быть обусловлено разницей расселения популяций в постледниковый период из разных рефугиумов [Санников, Петрова, 2012]. При этом полученные данные согласуются с предыдущими исследованиями в регионе [Vasilyeva et al., 2021; Chertov et al., 2022; Sboeva et al., 2022].
Таблица 2
Генетическая структура и дифференциация девяти популяций P. sylvestris [Genetic structure and differentiation of the nine populations of P. sylvestris ]
|
ISSR-праймер |
Нуклеотидная последовательность (5'→ 3') |
H T |
H S |
G ST |
|
CR-212 |
CTCTCTCTCTCTCTCTTG |
0.167 (0.012) |
0.122 (0.004) |
0.268 |
|
CR-215 |
CACACACACACAGT |
0.266 (0.022) |
0.161 (0.008) |
0.392 |
|
ISSR-1 |
ACACACACACACACACT |
0.157 (0.023) |
0.098 (0.010) |
0.377 |
|
M27 |
GAGAGAGAGAGAGAGAC |
0.226 (0.027) |
0.145 (0.012) |
0.360 |
|
X10 |
AGCAGCAGCAGCAGCAGCC |
0.229 (0.008) |
0.178 (0.007) |
0.221 |
|
На общую выборку |
0.205 (0.018) |
0.141 (0.008) |
0.315 |
Примечания: H T – ожидаемая доля гетерозиготных генотипов как мера общего генного разнообразия во всей популяции; H S – ожидаемая доля гетерозиготных генотипов в субпопуляции как мера ее внутрипопуляционного разнообразия или среднее выборочное генное разнообразие по всем локусам; G ST – доля межпопуляционного генетического разнообразия в общем разнообразии или показатель подразделенности популяций; в скобках даны стандартные отклонения.
На основе матрицы парных генетических расстояний (PhiPT) был проведен кластерный анализ методом Neighbor-joining и построена дендрограмма, отражающая степень сходства ISSR-профилей изученных популяций P. sylvestris (рис. 2). На дендрограмме изученные популяции были разделены на четыре кластера. Популяция Каслинского р-на выделилась в отдельную группу, что может быть связано с особенностями её расположения – на Аракульском Шихане, самом южном и высоком скальном массиве Среднего Урала.
Разделение популяций на четыре кластера подтверждается результатами анализа главных координат (PCoA), основанного на индексе PhiPT, рассчитанном с помощью пакета AMOVA (рис. 3).
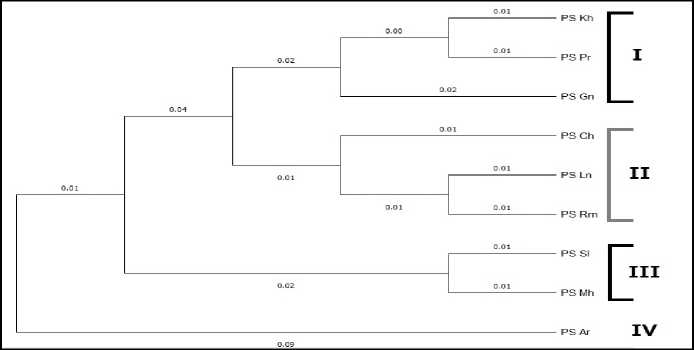
Рис. 2. Дендрограмма генетического сходства изученных популяций P. sylvestris , построенная на основе полиморфизма ISSR-фрагментов методом Neighbor-joining:
I, II, III, IV – номера кластеров; цифрами над ветвями указаны генетические расстояния
[Dendrogram of genetic similarity of studied populations of P. sylvestris , built on the basis of polymorphism of ISSR profiles by Neighbor-joining method:
I, II, III, IV – cluster numbers; the numbers above the branches indicate genetic distances]
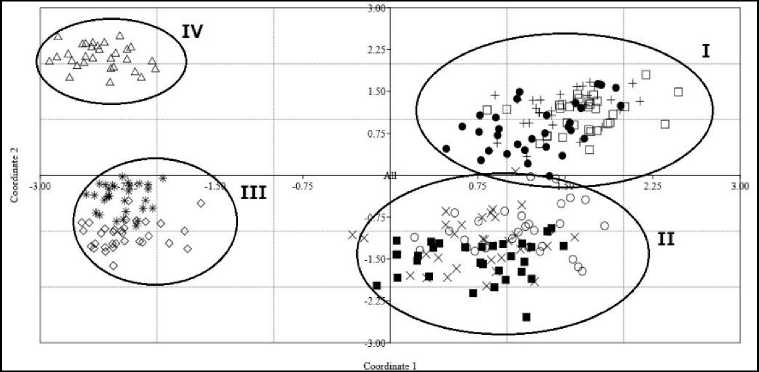
Рис. 3. Ординация изученных популяций P. sylvestris с использованием PCoA, полученного на основе матрицы генетических расстояний PhiPT:
точка – PS_Kh , плюс – PS_Gn , квадрат – PS_Pr , закрашенный квадрат – PS_Ln , крест – PS_Ch , круг – PS_Rm , ромб – PS_Sl , звёзда – PS_Mh , треугольник – PS_Ar ; I, II, III, IV – номера кластеров
[Ordination of the studied populations of P. sylvestris using PCoA, obtained on the basis of PhiPT matrix of genetic distances:
dot – PS_Kh , plus – PS_Gn , square – PS_Pr , fill square – PS_Ln , cross – PS_Ch , circle – PS_Rm , rhombus – PS_Sl , star – PS_Mh , triangle – PS_Ar; I, II, III, IV – cluster numbers]
При изучении популяций P. sylvesrtis их пространственная и генетическая структуры проверялась на соответствие модели «изоляция на расстоянии» с помощью корреляционного теста Мантела. Таким образом, попарное сравнение всех исследованных популяций выявило наличие средней положительной корреляции (R2 = 0,358) между географическим и генетическим расстоянием (PhiPT), что позволяет предположить, что существенная часть различий между популяциями обусловлена расстоянием между ними.
В целом изученные популяции дифференцируются согласно их географическому расположению, но в то же время их дифференциация обусловлена и другими факторами. Одним из таких факторов может быть история расселения популяций P. sylvestris. Согласно исследованию С.Н. Санникова с коллегами, уральские популяции заселялись преимущественно из Южноуральского рефугиума, но кроме того, заселение происходило из Балканского рефугиума и рефугиумов второго порядка в Южной Сибири [Санников и др., 2014; Санников и др., 2020]. Другим фактором может быть сложный рельеф региона исследо- вания. Так, южные популяции (PS_Ar, PS_Mh, PS_Sl), обособившиеся в отдельные кластеры произрастают на высотах 338, 291 и 302 м над ур. м. соответственно, тогда как остальные изученные популяции произрастают на высотах ниже 200 м над ур. м.
Результаты данного исследования генетической структуры и дифференциации природных популяций сосны обыкновенной на Урале позволяют рекомендовать для сохранения генетического разнообразия вида в регионе исследований по одной популяции из каждого кластера: из I – популяцию из Кишерского р-на (PS_Pr), из II – популяцию из Чердынского р-на (PS_Ch) Пермского края, из III – популяцию из Са-лаватского р-на (PS_Sl) Республики Башкортостан и из IV – популяцию из Каслинского р-на Челябинской обл.
Заключение
Изученные 9 популяций Pinus sylvestris L., расположенные на Урале, характеризуются средним уровнем генетического разнообразия ( P 95 = 0.886; I = 0.224; H E = 0.141; n e = 1.301), но в популяциях Южного Урала отмечено существенное его снижение у популяций из Мечетлинского ( PS_Mh ) и Салаватского р-нов ( PS_Sl ) Республики Башкортостан, ожидаемая гетерозиготность ( H E ) 0.091 и 0.071 соответственно.
Установлено, что изученные популяции P. sylvestris дифференцированы в средней степени ( G ST = 0.315; PhiPT = 0.371), но менее, чем популяции Восточно-Европейской равнины ( G ST = 0.488; PhiPT = 0.371). Популяции сосны обыкновенной в регионе исследования образуют четыре кластера, в целом соответствующих их географическому расположению и отражающих их расселение из Южноуральского рефугиума. Корреляционный тест Мантела позволил установить положительную корреляцию между генетическими и географическими расстояниями, что дает возможность предположить, что существенная часть различий между популяциями обусловлена расстоянием между ними, но также не стоит исключать влияния рельефа местности, истории расселения популяций и интенсивности лесозаготовок. Знания о генетической дифференциации сосны обыкновенной позволят отобрать популяции для изучения БАВ и дальнейшей молекулярно-генетической идентификации популяций сосны обыкновенной. На основании полученных данных отобраны по одной популяции из каждого установленного кластера для сохранения генетических ресурсов сосны обыкновенной на Урале.
Список литературы Анализ генетического разнообразия, структуры и дифференциации популяций Pinus sylvestris L. на Урале
- Боронникова С.В. и др. Молекулярно-генетическая идентификация в лесном хозяйстве с использованием геномных технологий // Бюллетень науки и практики. 2018. Т. 4, № 7. С. 26-33.
- Видякин А.И. и др. Генетическая изменчивость, структура и дифференциация популяций сосны обыкновенной (Pinus sylvestris L.) на северо-востоке Русской равнины по данным молекулярно-генетического анализа // Генетика. 2015. Т. 51, № 12. С. 1401-1409.
- Видякин А.И. и др. Распространение гаплотипов митохондриальной ДНК в популяциях сосны обыкновенной (Pinus sylvestris L.) на севере Европейской России // Генетика. 2012. Т. 48, № 12. С. 14401444.
- Потокина Е. К., Александрова Т.Г. Коэффициенты генетической оригинальности образцов коллекции вики посевной (Vicia sativa L.) по результатам молекулярного маркирования // Генетика. 2008. Т. 44, № 11. С. 1508-1516
- Пришнивская Я.В. и др. Внутривидовое генетическое разнообразие популяций двух видов древесных растений Пермского края // Бюллетень науки и практики. 2019. Т. 5, № 4. С. 58-68.
- Рябухина М.В. и др. Генетическое разнообразие популяций сосны обыкновенной Pinus sylvestris L. // Теоретическая и прикладная экология. 2019. № 3. С. 66-71.
- Санников С.Н., Петрова И.В. Филогеногеография и генотаксономия популяций вида Pinus sylvestris L. // Экология. 2012. № 4. С. 252-260.
- Санников С.Н. и др. Выявление системы плейстоценовых рефугиумов Pinus sylvestris L. в южной маргинальной зоне ареала // Экология. 2014. № 3. С. 174-181.
- Санников С.Н. и др. Поиск и выявление системы плейстоценовых рефугиумов вида Pinus sylvestris L. // Экология. 2020. № 3. С. 181-189.
- Сбоева Я.В. Оценка состояния генофондов популяций Pinus sylvestris L. на востоке и северо-востоке Восточно-Европейской равнины // Вестник Пермского университета. Сер. Биология. 2023. Вып. 4. С. 375-384.
- Чертов Н.В. и др. Молекулярно-генетическая идентификация популяций Pinus sylvestris L. и Larix sibirica Ledeb. в Пермском крае с использованием SNP-маркеров // Бюллетень науки и практики. 2020. Т. 6, № 12. С. 14-22.Art. 1278.
- Dering M. et al. Tertiary remnants and Holocene colonizers: Genetic structure and phylogeography of Scots pine reveal higher genetic diversity in young boreal than in relict Mediterranean populations and a dual colonization of Fennoscandia // Diversity and distributions. 2017. Vol. 23, № 5. P. 540-555.
- Floran V., Sestras R.E., Gil M.R. Organelle genetic diversity and phylogeography of Scots pine (Pinus sylvestris L.) // Notulae Botanicae Horti Agrobotanici Cluj-Napoca. 2011. Vol. 39, № 1. P. 317-322.
- Frankham R. How closely does genetic diversity in finite populations conform to predictions of neutral theory? Large deficits in regions of low recombination // Heredity. 2012. Vol. 108, № 3. P. 167-178.
- Hammer 0., Harper D.A.T., Paul D.R. Past: Paleontological statistics software package for education and data analysis // Palaeontol. Electron. 2001. Vol. 4, № 1. P. 1-9.
- Hebda A., Wojkiewicz B., Wachowiak W. Genetic characteristics of Scots pine in Poland and reference populations based on nuclear and chloroplast microsatellite markers // Silva Fennica. 2017. Vol. 51, № 2. P. 1-17.
- Hogberg P. et al. Large-scale forest girdling shows that current photosynthesis drives soil respiration // Nature. 2001. Vol. 411, № 683. P. 789-792.
- Kalendar R., Muterko A., Boronnikova S. Retrotransposable elements: DNA fingerprinting and the assessment of genetic diversity // Methods Mol. Biol. 2021. Vol. 2222. P. 263-286.
- Kalendar R. et al. Palindromic sequence-targeted (PST) PCR: A rapid and efficient method for high-throughput gene characterization and genome walking // Sci. Rep. 2019. Vol. 9, № 1. Art. 17707.
- Kavaliauskas D., Danusevicius D., Baliuckas V. New insight into genetic structure and diversity of Scots pine (Pinus sylvestris L.) populations in Lithuania based on nuclear, chloroplast and mitochondrial DNA markers // Forests. 2022. Vol. 13. Art. 1179.
- Khanova E. et al. Genetic and selection assessment of the Scots pine (Pinus sylvestris L.) in forest seed orchards // Wood Res. 2020. Vol. 65. P. 283-292.
- Kumar S. et al. Mega x: Molecular evolutionary genetics analysis across computing platforms // Mol. Bi-ol. Evol. 2018. Vol. 35, № 6. P. 1547-1549.
- Linden A. et al. Contrasting effects of increased carbon input on boreal SOM decomposition with and without presence of living root system of Pinus sylvestris L. // Plant and soil. 2014. Vol. 377. P. 145-158.
- Liu X. et al. Abietic acid suppresses non-small-cell lung cancer cell growth via blocking IKKP/NF-kB signaling // OncoTargets and therapy. 2019. P. 4825-4837.
- Nei M. Molecular population genetics and evolution. Amsterdam: North-Holland Publishing Company, 1975. 228 p.
- Nei M., Li W.H. Mathematical model for studying genetic variation in terms of restriction endonucleases // Proc. Natl. Acad. Sci. USA. 1979. Vol. 76, № 10. P. 5269-5273.
- O'Grady J.J. et al. Realistic levels of inbreeding depression strongly affect extinction risk in wild populations // Biological conservation. 2006. Vol. 133, № 1. P. 42-51.
- Pan Y. et al. A large and persistent carbon sink in the world's forests // Science. 2011. Vol. 333. P. 988993.
- Pazouki L. et al. Large within-population genetic diversity of the widespread conifer Pinus sylvestris at its soil fertility limit characterized by nuclear and chloroplast microsatellite markers // European journal of forest research. 2016. Vol. 135. P. 161-177.
- Peakall R.O.D., Smouse P.E. Genalex 6: Genetic analysis in Excel. Population genetic software for teaching and research // Mol. Ecol. Notes. 2006. Vol. 6, № 1. P. 288-295.
- Rogers S.O., Bendich A.J. Extraction of DNA from milligram amounts of fresh, herbarium and mummified plant tissues // Plant molecular biology. 1985. Vol. 5, № 2. P. 69-76.
- Sboeva Y. et al. Genetic Diversity, Structure, and Differentiation of Pinus sylvestris L. Populations in the East European Plain and the Middle Urals // Forests. 2022. Vol. 13, № 11. Art. 1798.
- Spielman D., Brook B.W., Frankham R. Most species are not driven to extinction before genetic factors impact them // Proceedings of the National Academy of Sciences. 2004. Vol. 101, № 42. P. 15261-15264.
- Toth E.G. et al. High genetic diversity and distinct origin of recently fragmented Scots pine (Pinus sylvestris L.) populations along the Carpathians and the Pannonian Basin // Tree Genetics & Genomes. 2017. Vol. 13. P. 1-2.
- Vasilyeva Y. et al. Genetic Structure, Differentiation and Originality of Pinus sylvestris L. Populations in the East of the East European Plain // Forests. 2021. Vol. 12, № 8. Art. 999.
- Yeh F.C. et al. POPGENE, the Microsoft Windows-based user-friendly software for population genetic analysis of co-dominant and dominant markers and quantitative traits // Dept. Renewable Resources, University of Alberta, Edmonton, Canada. 1996. Vol. 238. P. 1-29.
- Zietkiewicz E., Rafalski A., Labuda D. Genome fingerprinting by simple sequence repeat (SSR)-anchored polymerase chain reaction amplification // Genomics. 1994. Vol. 20, № 2. P. 176-183.
