Болезнь Вильсона-Коновалова: обзор литературы и случай из практики

Автор: Юсупов Фуркат Абдулахатович, Юлдашев Акмал Акбарович
Журнал: Бюллетень науки и практики @bulletennauki
Рубрика: Медицинские науки
Статья в выпуске: 3 т.9, 2023 года.
Бесплатный доступ
Гепатолентикулярная дегенерация (болезнь Вильсона-Коновалова) является тяжелым наследственным заболеванием, при котором нарушается метаболизм меди и характеризуется патологическим накоплением меди в головном мозге, печени и других органах. Болезнь вызвана мутацией в АТФазной медьтранспортирующей бета-полипептида (ATP7B), который кодирует трансмембранную АТФазу, транспортирующую медь, что приводит к накоплению меди в организме. Клиническое течение может варьироваться по типу и тяжести симптомов, но прогрессирующее течение со стороны нервной системы и печени является общей особенностью. Пациенты также могут страдать кроме неврологических расстройств и психическими симптомами. Гепатолентикулярная дегенерация диагностируется с использованием диагностических алгоритмов, которые включают анамнез заболевания, клинические симптомы и признаки, показатели метаболизма меди и анализ ДНК ATP7B. Доступные методы лечения включают хелатную терапию и соли цинка, которые устраняет дальнейшее накопление меди. Кроме того, в отдельных случаях показана трансплантация печени. При ранней диагностике и лечении прогноз хороший; однако важным вопросом является диагностика пациентов до появления серьезных симптомов. В работе сделан обзор последних данных по болезни Вильсона-Коновалова с подробным описанием этиологии, патогенеза, макроскопические и микроскопические изменении в органах мишени, неврологические проявления, диагностика, дифференциальная диагностика, новые методы лечения, прогноз и осложнения. Приведен клинический случай болезнь Вильсона-Коновалова у 28-летнего пациента с печеночными и экстрапирамидными проявлениями.
Болезнь вильсона-коновалова, гепатолентикулярная дегенерация, медь, церулоплазмин, экстрапирамидные расстройства
Короткий адрес: https://sciup.org/14127163
IDR: 14127163 | УДК: 616.831: | DOI: 10.33619/2414-2948/88/22
Wilson-Konovalov disease: literature review and case study
Wilson-Konovalov disease (hepatolenticular degeneration) is a heavy potentially treatable hereditary disorder of copper metabolism, which is characterized by pathological accumulation of copper. The disease is caused by mutations in ATPase copper transporting beta polypeptide (ATP7B), which encodes the transmembrane ATPase transporting copper, which leads to a violation of copper homeostasis and copper overload in the liver, brain and other organs. The clinical course may vary by type and severity of symptoms, but progressive liver disease is a common feature. Patients may also suffer from neurological disorders and mental symptoms. Hepatolenticular degeneration is diagnosed using diagnostic algorithms that include clinical symptoms and signs, indicators of copper metabolism and ATP7B DNA analysis. Available treatments include chelation therapy and zinc salts, which eliminate copper overload by various mechanisms. In addition, liver transplantation is indicated in some cases. With early diagnosis and treatment, the prognosis is good; however, an important issue is the diagnosis of patients before the appearance of serious symptoms. The paper reviews the latest data on Wilson-Konovalov disease with a detailed description of the etiology, pathogenesis, macroscopic and microscopic pictures of the disease, neurological manifestations, diagnosis, differential diagnosis, new treatment methods, prognosis and complications. A clinical case of Wilson - Konovalov disease in a 28-year-old patient with extrapyramidal manifestations is presented.
Текст обзорной статьи Болезнь Вильсона-Коновалова: обзор литературы и случай из практики
Бюллетень науки и практики / Bulletin of Science and Practice Т. 9. №3. 2023
УДК 616.831: 616.36
Болезнь Вильсона-Коновалова представляет собой один из классических примеров болезней при котором поражается преимущественно головной мозг и печень. Болезнь имеет разные названии: гепатолентикулярная дегенерация, гепатоцеребральная дистрофия, болезнь Вестфаля-Вильсона, болезнь Вестфаля-Вильсона-Коновалова. Болезнь Вильсона-Коновалова это одна из наиболее изученных наследственных форм экстрапирамидной патологии, связана с нарушением обмена церулоплазмина — белка плазмы крови, содержащий медь, и синтезируется в печени. Морфологически выявляется отложение меди преимущественно в подкорковых ганглиях (n. Lenticularis), коре полушарий головного мозга, мозжечке, а также в печени, селезенке, радужке и хрусталике. В пораженных органах развиваются очаги размягчения и склерозирования. Тип наследования: аутосомно-рецессивный, встречается одинаково часто у мужчин и женщин. Болезнь представляет широкий интерес для неврологов, гепатологов и гастроэнтерологов. Выделяют брюшная форма (цирроз, гепатит, острая печеночная недостаточность), церебральная форма (экстрапирамидная, церебеллярная, псевдобульбарная, судороги, когнитивные и психические нарушения), смешанная форма (поражения нервной системы, печени, почек, глаз, суставов, сердца, эндокринной системы).
Целью данного обзора явилось изучить современные данные об этиологии, патогенеза, клинической картины, диагностики, дифференциальной диагностики, лечении, осложнений и прогноз при болезни Вильсона-Коновалова (БВК), а также привести случай из практики.
Болезнь Вильсона вызвана одной из нескольких мутаций в гене АТФазный медьтранспортирующий бета-полипептид (ATP7B), присутствующем на 13 хромосоме, которая контролирует белковый транспортер, ответственный за выведение избытка меди из организма через желчь. Около 10% случаев мутация в гене может и не обнаруживаться. Транспортер белка расположен в сети комплекса Гольджи печени и головного мозга. Основной путь выведения меди (95%) проходит через печень. Этот избыток меди сначала накапливается в печени, а затем попадает в кровь, центральную нервную систему (ЦНС) и в другие органы [1].
Избыток меди приводит к образованию свободных радикалов, которые вызывают окисление жизненно важных белков и липидов. Ранние изменения, как правило, происходят в митохондрии, ядрах и пероксисомах. При болезни Вильсона нарушается механизм выделения меди, в результате чего медь накапливается в печени и попадает в кровь, где начинает накапливаться в других органах и тканях, таких как гипоталамус, скорлупа и кора больших полушарий, почки и роговица. Медь является переходным металлом, и избыточное содержание меди приводит к образованию токсичной гидроксильной группы и усилению окислительного стресса в клетках. Этот окислительный стресс повреждает клетки и приводит к клиническим проявлениям, а именно к печеночной недостаточности, поведенческим нарушениям, расстроствам движений и появление кольца Кайзера-Флейшера в роговице [2].
Медь необходима организму преимущественно в качестве кофактора для некоторых ферментов, таких как церулоплазмин, оксидаза цитохрома с, бета-гидроксилаза допамина, супероксиддисмутаза и тирозиназа. Медь поступает в организм через пищеварительный тракт с помощью белка-переносчика в клетках тонкой кишки, медного мембранного транспортера 1 (Ctr1; SLC31A1). Этот транспортер помогает переносить медь внутри клеток, где часть меди связана с металлопротеином, а часть переносится ATOX1 к органелле, известной как сеть комплекса Гольджи. В ответ на повышение уровня меди фермент, называемый ATP7A, высвобождает медь в воротную вену в печень. Клетки печени несут белок CMT1 и металлопротеин, а затем ATOX1 связывает его внутри клетки. Оказавшись здесь, ATP7B связывает медь с церулоплазмином и высвобождает ее в кровоток, удаляя избыток меди, выделяя ее в желчь. Обе функции ATP7B нарушены при болезни Вильсона-Коновалова. Медь накапливается в печени, и церулоплазмин выделяется в форме, в которой не хватает меди, и быстро разлагается в кровотоке. Когда уровень меди в печени подавляет белки, которые обычно ее связывают, это приводит к окислительному повреждению в результате процесса, известного как химия Фентона. Это повреждение приводит к хроническому активному гепатиту, фиброзу и циррозу печени. Печень выделяет в кровоток медь, которая не связана с церулоплазмином. Эта свободная медь осаждается по всему организму, особенно в почках, глазах и головном мозге. В головном мозге медь откладывается в базальных ганглиях, путамене и бледном шаре (т. е., чечевицеобразное ядро); эти области участвуют в координации движений и нейрокогнитивных процессах, таких как стимулирование регуляции настроения. Повреждение этих областей вызывает нервно-психические симптомы, наблюдаемые при болезни Вильсона-Коновалова [3].
90–95% циркулирующих меди в сыворотке крови переносится церрулоплазмином. Церрулоплазмин синтезируется в печени гепатоцитами в двух формах, связанный в виде холоцеррулоплазмин а связанные аппоцеррулоплазмин. В одной молекуле этого белка содержится 6–7 ионов меди. В течение всей жизни в норме уровень церрулоплазмина остается стабильным за исключением неонатального периода, и у беременных. Уровень этого белка может повышаться при воспалительных процесса в организме. На Рисунке 1. представлено строение церрулоплазмина, а на Рисунке 2 — метаболизм меди и его роль в патогенезе гепатолентикулярной дегенерации.
Рисунок 1. Строение церрулоплазмина под микроскопом [4]
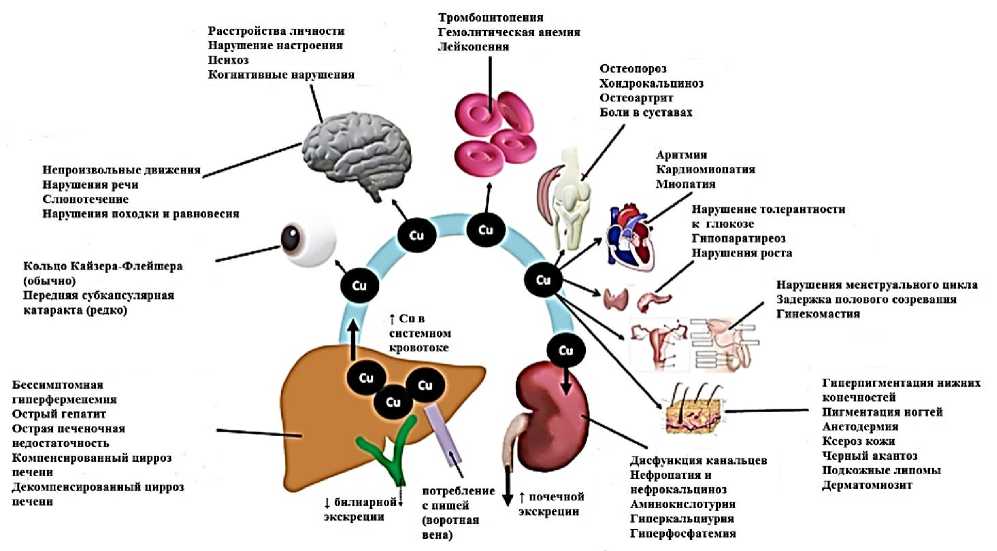
Рисунок 2. Метаболизм меди и его роль в патогенезе болезни Вильсона [5]
Гистология. Световая микроскопия может показать ранние изменения, включая умеренную жировую инфильтрацию и отложение гликогена в гепатоцитах. Гистологические особенности могут напоминать хронический активный гепатит. Несмотря на то, что уровень меди в печени повышен, на ранней стадии медь находится в цитоплазме и может не проявляться при окрашивании родамином. Т. С. Гулевская и соавторы изучили 15 умерших с БВК и обнаружили как макроскопические, так и микроскопические изменения. Макроскопические изменения наиболее заметно были в скорлупе и ограде, меньшей степени в хвостатом ядре и бледном шаре. Они были уменьшены в размере, мозговой ткан в этих областях имел губчатую структуру. Микроскопические изменения были грубыми и были обнаружены различных областях. При этом изменения были обнаружены во всех структурных элементах: сосудах, нейронах, глиальных элементах. Сосудистые изменения затрагивала преимущественно в микроциркуляторном русле. Эти изменения характеризовались как плазматические пропитывания, гиалиноз и фиброз стенки сосудов, стазам и дистонией.
Клинические проявления. Поскольку болезнь Вильсона-Коновалова является наследственным заболеванием, пациенты могут иметь положительный семейный анамнез. Описанный возраст начала болезни от 2 до 60 лет. Не менее чем четверти случаев заболевание не диагностируется, либо диагностируется недопустимо поздно. Суточное потребление меди с пищей составляет около 1 мг. Около 60 мг поглощенной меди (0,6 мг/день) абсорбируется в кишечнике. 0,35 мг меди в день выводится через кожу. 0,2 мг меди вдень экскретируется с желчью («регуляторная» медь). 0,05 мг меди в день выводится с мочой. Пациенты могут жаловаться на боли в животе, желтуху, слабость, изменения личности, депрессию, мигренозные головные боли, бессонницу, судороги и двигательного расстройства в виде хореи или хореоатетоза, гемибаллизм. Примерно у 30–50% пациентов будут наблюдаться нервнопсихические симптомы, включая асимметричный тремор. Другие симптомы могут включать слюнотечение, атаксию, изменения личности, маскообразные черты лица и неуклюжесть.
По данным [6] Valentina Medici и соавторов выделяют 3 фенотипическе проявления: печеночные (40%); неврологические (40%); бессимптомные (20%).
Бюллетень науки и практики / Bulletin of Science and Practice Т. 9. №3. 2023
В то же время авторы утверждает, что в реальной клинической практике в большинстве случаев встречается сочетанные поражения головного мозга и печени. Существует две формы течения заболевания:
-
1. Острое течение, при этом болезнь манифестируется в раннем детском возрасте, протекает молниеносно с высоким процентом летальности.
-
2. Хроническое течение, при котором симптоматика развивается медленно и часто с признаками поражения печени с последующим нарастанием неврологической симптоматики.
Клиническая классификация гепатолентикулярной дегенерации (по Н. В. Коновалову): «Брюшная» форма (доневрологическая); аритмогиперкинетическая (ранняя) форма; дрожательная форма; экстрапирамидно-корковая форма.
При объективном осмотре у пациента могут быть признаки гепатоспленомегалии, изолированной спленомегалии или, если заболевание прогрессировало до цирроза, также могут быть заметны признаки хронического заболевания печени. Осмотр глаз может выявить желтуху склер и исследование щелевой лампы на наличие колец Кайзера-Флейшера (KФ) на роговице (обратите внимание, что единственным другим заболеванием с кольцами KФ является первичный билиарный цирроз). Другие особенности болезни Вильсона могут включать наличие двигательных расстройств, трудности с речью, лица, похожие на маски, спастичность, ригидность мышц и кожные проявления лунно-голубых (синеватое обесцвечивание у основания ногтей). Поражение скелета очень часто встречается и напоминает преждевременный остеоартрит. Артропатия обычно поражает осевой скелет и позвоночник. Гемолитическая анемия наблюдается у 10–15% пациентов и обусловлена лизисом эритроцитов высокой концентрацией меди. Когда врачи сталкиваются с молодым пациентом с нарушением функции печени, гемолитической анемией и нормальным уровнем щелочной фосфатазы, следует заподозрить гепатолентикулярной дегенерации. Почечные симптомы похожи на синдром Фанкони и мочекаменную болезнь. Ниже в Таблице 1 подробно описан основные симптомы со стороны органов и систем.
Таблица 1 КЛИНИЧЕСКИЕ ПРОЯВЛЕНИЯ БОЛЕЗНЬ ВИЛЬСОНА-КОНОВАЛОВА [7]
|
Мишень |
Симптомы |
|
Неврологические проявления |
Дистония, тремор, дизартрия, дисфагия, акинетико-ригидный синдром, хорея |
|
Психиатрическая проявления |
Поведенческие изменения, депрессия, тревога, психоз, неуспеваемость в школе, сексуальные нарушения |
|
Со стороны печени |
Гепатомегалия, желтуха, боль в правом подреберье, астения, повышение уровня трансаминаз, признаки острой печеночной дисфункции, цирроз печени (компенсированный и декомпенсированный), стеатоз |
|
Со стороны органов глаз |
Кольцо Кайзера-Флейшера, катаракта |
|
Гематологические проявления |
Гемолитическая анемия, коагулопатия, тромбопения |
|
Почечные проявления |
Острая почечная недостаточность, нефролитиаз, мочекаменная болезнь, уремия |
|
Проявления со стороны опорнодвигательного аппарата |
Артропатия, мышечная слабость |
|
Другие проявления |
Проявления со стороны сердечно-сосудистой системы, панкреатит, гипопаратиреоз |
Диагностический поиск. Если подозрение на болезнь Вильсона-Коновалова высокое, необходимо проверить уровень церулоплазмина. Это будет менее 20 мг/дл (в норме от 20 мг/дл

Бюллетень науки и практики / Bulletin of Science and Practice Т. 9. №3. 2023 до 40 мг/дл). Уровень меди в моче будет повышен более чем на 100 мкг/дл. Этих двух лабораторных результатов с кольцами Кайзера-Флейшера обычно достаточно для постановки диагноза, но, если есть возможность для биопсии печени на содержание меди в гепатоцитах; это наиболее точный метод для окончательного установления диагноза. Примечание: низкий уровень церулоплазмина может наблюдаться при любом расстройстве сочетающиеся дефицитом белка. МРТ голоного мозга помогает определить наличие очаги поражения (отложения меди особенно области базальных ганглиев) поражения головного мозга. Традиционные печеночные тесты показывает повышенный уровень АСТ и АЛТ [8, 9]. Болезнь Вильсона-Коновалова следует заподозрить, если присутствуют симптомы, соответствующие этому заболеванию, или если у родственника было обнаружено это заболевание. У большинства из них были слегка аномальные показатели печеночных тестов и повышенный уровень аспартаттрансаминазы, аланиновой трансаминазы и билирубина. Если повреждение печени является значительным, альбумин снижается в результате неспособности поврежденных клеток печени вырабатывать этот белок; аналогично, протромбиновое время увеличивается, поскольку печень не вырабатывает белки, известные как факторы свертывания крови. Уровни щелочной фосфатазы низкие у пациентов с острой печеночной недостаточностью вследствие гепатолентикулярной дегенерации. При наличии неврологических симптомов МРТ головного мозга может показать гиперинтенсивность в базальных ганглиях в режиме T2. МРТ может продемонстрировать характерный рисунок «лицо гигантской панды». ЭКГ может выявить гипертрофию желудочков, аритмии и неспецифические изменения в Т-волнах и сегментах ST. Наличие колец Кайзера-Флейшера при наличии нервно-психических симптомов наводит на мысль о болезни Вильсона. Полностью надежного теста на болезнь Вильсона не существует, но уровни церулоплазмина и меди в крови, а также меди, выделяемой с мочой в течение 24-часового периода, используются для формирования впечатления о количестве меди в организме. Золотым стандартом является биопсия печени. Родственники первой и второй степени нуждаются в обследовании на болезнь Вильсона.
Таблица 2
ЛЕЙПЦИГСКАЯ КОЛИЧЕСТВЕННАЯ ШКАЛА ДЛЯ ДИАГНОСТИКИ
БОЛЕЗНИ ВИЛЬСОНА-КОНОВАЛОВА
(8th International Meeting on Wilson’s disease, Leipzig, 2001)
|
Признак |
Выраженность |
Балл |
|
Характерные клинические признаки |
||
|
Кольца Кайзера-Флейшера |
Есть |
2 |
|
на роговице глаза |
Нет |
0 |
|
Неврологические симптомы |
Тяжелые |
2 |
|
или характерные проявления |
Легкие |
1 |
|
при МРТ головного мозга |
Отсутствует |
0 |
|
Концентрация |
Нормальная (> 0,2 г/л или (> 200 мг/л) |
2 |
|
церулоплазмина сыворотки |
0,1–0,2 г/л или 100–200 мг/л |
1 |
|
<0,1 г/л или <100 мг/л |
0 |
|
|
Гемолитическая анемия с |
Имеются |
1 |
|
отрицательно пробой Кумбса |
Отсутствует |
0 |
|
Другие методы исследования |
||
|
Содержание меди в печени (при отсутствии холестаза) |
В 5 раз выше верхней границы нормы (> 4 мкмоль/г или > 250 мкг/г) |
2 |
|
0,8–4 мкмоль/г или 50–250 мкг/г |
1 |
|
Бюллетень науки и практики / Bulletin of Science and Practice Т. 9. №3. 2023
|
Признак |
Выраженность |
Балл |
|
Нормальное (<0,8 мкомоль/г или <50 мкг/г) |
-1 |
|
|
Наличие роданин-позитивных гранул (при отсутствии возможности количественного определения меди) |
1 |
|
|
Экскреция меди с мочой (при |
Нормальная (<0,9 мкмоль/сут. × 1,73 м2 или <57 |
0 |
|
отсутствии острого гепатита) |
мкг/сут. × 1,73 м2) Выше верхней границы нормы в 2 раза и меньше |
1 |
|
Более чем в 2 раза выше верхней границы нормы |
2 |
|
|
Нормальная, но повышается более чем в 5 раз выше верхней границы нормы при приеме d-пеницилламина |
2 |
|
|
Молекулярно-генетическая |
Мутации в 2-х хромосомах |
4 |
|
диагностика |
Мутации в 1 хромосоме |
1 |
|
Дефекты мутаций не выявлены |
0 |
|
|
Интерпретация результата |
||
|
Диагноз установлен |
≥ 4 |
|
|
Диагноз сомнителен, необходимо исследование большего числа показателей |
3 |
|
|
Диагноз маловероятен |
≤ 2 |
|
Лечение. Основной терапией болезни Вильсона-Коновалова является хелатная терапия (ХТ) меди пенициламином и триентином. Триентин предпочтительнее из-за меньшего количества побочных эффектов. Пероральный цинк также может быть дан, поскольку он конкурирует за поглощение с медью при транспортировке металлических ионов. Жизненно важно информировать пациента о побочных эффектах хронической хелатной терапии, которые могут усугубить симптомы. D-пеницилламин можно применять во время беременности и не представляет никакого риска для плода. Если у пациента развивается цирроз печени и связанное с ним осложнение, при рецидивирующем варикозном кровотечении может быть предложен трансюгулярный внутрипеченочный портосистемный шунт. Трансплантация печени является лечебной [10–12].
При мышечной ригидности, спастичности и особенностях паркинсонии могут быть использованы баклофен, антихолинергические препараты (тригексифенидил), антагонисты ГАМК и леводопа. Трансплантация печени, по-видимому, была полезна для улучшения неврологической дисфункции у некоторых пациентов, которые неадекватно реагировали на медикаментозную терапию во французском исследовании [13].
Рекомендуется диета с низким содержанием продуктов, содержащих медь, с отказом от грибов, шоколада, орехов, сухофруктов, печени и моллюсков. Физиотерапия и трудотерапия полезны при неврологической форме заболевания. Лечение хелатированием меди занимает до шести месяцев, чтобы начать работать, и эти методы лечения могут помочь справиться с атаксией, дистонией и тремором, а также предотвратить контрактуры, которые могут возникнуть в результате дистонии. Современные фармакологические методы лечения в значительной степени не помогли спасти гомеостаз меди у пациентов с острой печеночной недостаточностью, в результате чего ХТ остается единственным жизнеспособным вариантом лечения. Недавнее исследование с использованием крысиной модели LEC БВК предложило дополнительный вариант для таких пациентов, называемый метанобактином, пептидом, продуцируемым Methylosinus trichosporium , с чрезвычайно высоким сродством к меди [20].
Краткосрочное лечение метанобактином (МБ) эффективно обратило вспять острое повреждение печени из-за накопления меди. Этот благотворный эффект был связан с удалением внутриклеточного меди, в частности из митохондрий. Интересно, что обычные хелаторы меди пеницилламин и тетратиомолибдат не смогли очистить токсичный металл от запасов митохондрий. Как следствие, лечение МБ предотвратило гибель гепатоцитов и последующую печеночную недостаточность, удлинив продолжительность жизни крысы LEC. Поэтому этот пептид, по-видимому, является потенциальным терапевтическим средством при острой форме гепатолентикулярной дегенерации. Результаты другого недавнего исследования показали, что агонист Х-рецептора печени (LXR)/ретиноидного Х-рецептора может быть использован для борьбы с токсичностью Cu при БВК [14, 15] хелатирования меди. Тщательное исследование транскрипционных и метаболических изменений в образцах от пациентов с гепатоцеребральной дегенерацией и мышей Atp7b выявило нарушение регуляции LXR как одно из ключевых событий в патогенезе гепатолентикулярной дегенерации [16].
Дифференциальная диагностика. Дифференциальная диагностика включает состояния, вызывающие хронический активный гепатит, гемолитические анемии, вызванные различными причинами. Нейропсихиатрические расстройства, которые могут быть ошибочно приняты, включают различные типы синдромов Паркинсона, нейродегенерацию, связанную с дефицитом пантотенаткиназы (накопление железа), синдромы нейроакантоцитоза и болезнь Хантингтона, и это лишь некоторые из них. Эти состояния могут сопровождаться изменениями личности, ригидностью, дистонией и двигательными расстройствами.
Стадии болезни Вильсона:
Стадия 1: начальное накопление меди в печени
Стадия 2: Острое перераспределение меди в печени с последующим высвобождением в системный кровоток.
Стадия 3: Хроническое накопление меди во внепеченочных тканях, включая головной мозг.
Стадия 4: Применение хелатотерапии для восстановления баланса меди
Прогноз. Для прогнозирования были предложены различные системы оценки. Некоторые из них включают уровни АСТ, билирубина, протромбинового времени и т. д. При предъявлении [17].
Таблица 3
РАСЧЕТ ПРОГНОСТИЧЕСКОГО ИНДЕКСА ПРИ ГЕПАТОЦЕРЕБРАЛЬНОЙ ДИСТРОФИИ (Dhawan A. et al., 2005)
|
Показатель |
1 балл |
2 балла |
3 балла |
4 балла |
|
Общий билирубин сыворотки, мкМ/л |
100–150 |
151–200 |
201–300 |
>301 |
|
АсАТ (МЕ/мл) |
100–150 |
151–300 |
301–400 |
>400 |
|
МНО |
1,3–1,6 |
1,7–1,9 |
2,0–2,4 |
>2,4 |
|
Число лейкоцитов, × 109 /л |
6,8–8,3 |
8,4–10,3 |
10,4–15,3 |
>15,3 |
|
Альбумин, г/л |
34–44 |
25–33 |
21–24 |
<21 |
Сумма баллов ≥ 11 ассоциируется с высокой вероятностью смерти без пересадки печени.
Пациенты с прогностическим баллом 7 и более должны быть направлены на трансплантацию печени. Пациенты с таким показателем, как правило, умирают в течение 8 недель без лечения. После трансплантации печени прогноз хороший. Сообщалось о выживаемости 87% в возрасте 15 лет [17]. Клеточная терапия, так же, как и генная терапия, нацелена на печень, которая не экспрессирует функциональные ATP7B белок, направленный на восстановление гепатобилиарной экскреции меди [15, 18, 19]. Клеточная терапия при БВК представляется возможной, поскольку трансплантированные гепатоциты могут интегрироваться в паренхиму печени и восстанавливать нарушенные функции, включая транспорт меди в желчь. Модели болезни на животных, особенно крыса LEC, облегчили исследования по трансплантации клеток в БВК [15, 19]. Именно с помощью этой модели на животных было установлено, что для достижения достаточного удаления меди и терапевтической эффективности необходимо повторно заселить менее половины печени здоровыми гепатоцитами. Однако в этом исследовании не каждое животное, подвергнутое клеточной терапии, показало эквивалентные преимущества [5, 13]. Внепеченочная клеточная терапия с использованием инженерных приложений (то есть, трансплантация ткани печени в тонкую кишку или брюшную полость) в настоящее время считается недостаточной, поскольку для удаления меди требуется неповрежденная желчевыделительная система. Таким образом, в случае клеточной или генной терапии при БВК печень является первой мишенью, учитывая физиологическое ограничение ATP7B экспрессия в гепатоцитах, а также наличие механизмов для выведения меди из организма [20, 21]. К счастью, исследования трансплантации с использованием донорских клеток подтвердили способность к выведению желчи, что дает первый ключ к пониманию того, что транспорт билиарного меди является возможной мишенью для клеточной терапии при БВК [2, 11, 18]. HLC, полученные из IPSC, могут повторно заселять печень, но они поддерживают только некоторые функции гепатоцитов, которые ограничены стадиями, подобными эмбриональным [2, 22]. Поэтому пересаженные клетки могут размножаться в присутствии нативных клеток, имеющих низкую скорость пролиферации. Это происходит, когда нативные клетки печени имеют обширное повреждение ДНК (вызванное токсинами, ишемией и/или гепатэктомией). Очевидно, что эти режимы предварительной подготовки нежелательны в условиях существующего повреждения печени при БВК. Способность трансплантированных HLC выражать ATP7B была оценена в печени мышей LEC, у которых количество пересаженных клеток не увеличивалось в течение периода наблюдения в несколько месяцев [5, 12]. Таким образом, репопуляция печени в течение очень длительного периода подразумевает, что терапевтическая коррекция при БВК потребует значительного времени [23, 24]. Генная терапия направлена на исправление дефекта в нативных гепатоцитах путем предоставления здоровых копий ATP7B путем введения трансгена векторами, способными к неограниченной интеграции и/или сохранению в клетках. Доказательством принципа генной терапии при БВК послужила экспрессия аденовирусного и лентивирусного вектора ATP7B в печени моделей грызунов, которая позволила добиться временной коррекции экскреции меди и включения последнего в церулоплазмин [25, 26]. Недавно эти первоначальные наблюдения были подтверждены аденоассоциированным векторным серотипом 8 (AAV8), кодирующим человеческую кДНК ATP7B, помещенную под контроль специфичного для печени промотора α1-антитрипсина (AAV8-AAT-ATP7B). Экспрессия AAV8-AAT-ATP7B у мышей с дефицитом ATP7B приводила к снижению ферментов печени и восстановлению физиологической экскреции меди с желчью [21]. Это исследование показало прочную основу для будущих трансляционных исследований. Генная терапия, опосредованная AAV, должна позволятьATP7B будет постоянно экспрессироваться в печени пациента с болезнью Вильсона-Коновалова и, следовательно, устранит необходимость в пожизненном приеме препаратов, снижающих содержанию меди. С другой стороны, такие риски, как иммунный ответ, биогенез опухоли и плохая интеграция в поврежденные клетки [27] должны быть взвешены перед терапией переноса генов, опосредованной AAV, и будут важным компонентом репертуара методов лечения гепатолентикулярной дегенерации. Кроме того, по-прежнему требуются исследования, изучающие судьбу генетически модифицированных нативных клеток в печени при БВК [15, 26]. Новые методы лечения. Чтобы определить, будут ли и когда эти новые методы лечения пригодны для клинических испытаний, необходимо выполнить ряд ключевых требований. Во-первых, необходимо определить конкретную референтную популяцию для лечения. Вполне вероятно, что потенциальные кандидаты в эту популяцию будут включать бессимптомных или предсимптомных пациентов с ранним заболеванием, людей с печеночной недостаточностью или тяжелым неврологическим ухудшением, которым могло бы помочь быстрое удаление меди, а также лиц с тяжелой побочной реакцией на ранее существовавшую терапию [18]. Учитывая потенциальную задержку некоторых методов лечения до того, как они станут эффективными, и серьезные риски, связанные с приостановкой ранее существовавшего лечения у пациентов с БВК, представляется возможным рассмотреть комбинацию существующих лекарств и новых методов лечения для мобилизации меди для ранних клинических испытаний. Кроме того, будет важно определить соответствующие тесты и эффективные конечные точки, которые, в дополнение к клиническим параметрам, могут быть полезны для демонстрации терапевтической эффективности, учитывая, что мониторинг лекарств требует использования неинвазивных анализов и биомаркеров. В этом контексте открытие новых биомаркеров в БВК будет иметь большое значение для обеспечения нового эффективного лечения.
Клинический случай . Больной Н. 28 лет. Поступил в неврологическое отделение с жалобы на выраженный грубый тремор рук, захватывающий туловища, затруднения при самообслуживании по причине выраженного грубого тремора рук, неустойчивость при ходьбе, частые падения при ходьбе, нарушение речи, в виде нечеткой артикуляции речи, слюнотечение, диффузные головные боли, головокружение, плохой сон, общую слабость, быструю утомляемость. Анамнез заболевания: болеет больше 10 лет. Начало заболевания связывает с полученной сочетанной травмой в результате ДТП. В то время получал соответствующее лечение по м/ж, с улучшением. Спустя 5 лет начал отмечать дрожание левой руки, в начале особого внимания не обращал, но в динамике с течением времени интенсивность тремора усилилась и распространился в правую руку. По поводу чего неоднократно получал лечение, без особого эффекта. В течение последних нескольких лет присоединились нарушение речи, неустойчивость при ходьбе. Анамнез жизни больной 4 по счету в семье сын. Родился в сроке и рост нормально. Со слов пациента наследственность не отягощен. Объективный статус при поступлении: Кожные покровы и видимые слизистые оболочки обычной окраски, чистые, периферических отеков нет. Грудная клетка в акте дыхания участвует симметрично. При аускультации в легких дыхание везикулярное, хрипов нет. ЧД 18 в мин. Сердечные тоны приглушены, ритмичные. АД 120/80 мм. рт. ст. ЧСС 74 уд в мин. Язык влажный, чистый. Живот мягкий, при пальпации безболезненный. Печень не увеличен. Стул и диурез со слов регулярны. Неврологический статус: Сознание сохранено. Ориентирован во времени и месте. Память снижена на текущие события. Дизартрия. Глазные щели правильной формы, одинаковой ширины, зрачки OD=OS, округлой формы. Фотореакция живая. Движение глазных яблок в полном объеме. Лицо симметричное. Язык media. Мышечная сила в рамках допустимой нормы. Тонус мышц верхних конечностей относительно повышен по пластическому типу, (+) феномен зубчатого колеса. Сухожильные и периостальные рефлексы D=S, In=Sp. Чувствительных расстройств нет. (+) Паторефлекс Маринеску -Радовичи с обеих сторон. Менингеальных знаков нет. Пальценосовую пробу выполняет с мимопопаданием и интенцией с двух сторон. Отмечается грубый, сложный (тремор покоя и акционный тремор) верхних конечностей, тремор «бьющихся крыльев». В позе Ромберга неустойчив, пошатывается в стороны. Тазовые функции регулирует. Лабораторные исследования: Развернутый анализ крови от 24.01.2022 г. Гемоглобин: 150 г/л, Эритроциты 5,01×1012 ед./л , Гематокрит 43,2%, Лейкоциты 3,70×109 ед./л, СОЭ: 4 мм/ч, нейтрофилы: с/я 69,4%, Лимфоциты: 20,5%. Моноциты 0,25% Тромбоциты 88×109 л. ОАМ от 24.01.2022 г. Цвет желтый. Прозрачность — неполная. Удельный вес >1030. Белок — 0,30 г/л, Эпителий плоские
Бюллетень науки и практики / Bulletin of Science and Practice Т. 9. №3. 2023
0–1 клетки в поле зрения., лейкоциты — 0–1 в п/з. Серологическое исследования от 24.01.2022 г. Гепатит В, HВs Ag (кач) — отрицательный. Гепатит С Anti HCV сумм. (кач) отрицательный. Сифилис — отрицательный. ПЦР SARS Cov-2 — не обнаружено. Биохимические анализы: от 24.01.2022 г. Общий белок — 69,5 г/л, Креатинин — 100 мкмоль/л, мочевина 7,2 ммоль/л, холестерин общий 2,65 ммоль/л, АЛТ — 25,2 ед./л, АСТ — 28,8 ед./л. Бил. общий — 36,3 мкмоль/л, сывороточное железо — 27,4 мкмоль/л. Церулоплазмин от 08.02.2022 г.: 20 мг/л (норма 150–300), медь (моча) 08.02.2022 г.: 528,0 мкг/л (2–80), медь (кровь) от 08.02.2022 г. 321,0 мкг/л (575–1725). Инструментальные данные: МРТ головного мозга от 08.02.2022 г. в режимах FLAIR, T2, T1 и диффузно-взвешенных изображениях (DWI), в аксиальной коронарной и сагитальной плоскостях (Рисунок 3).
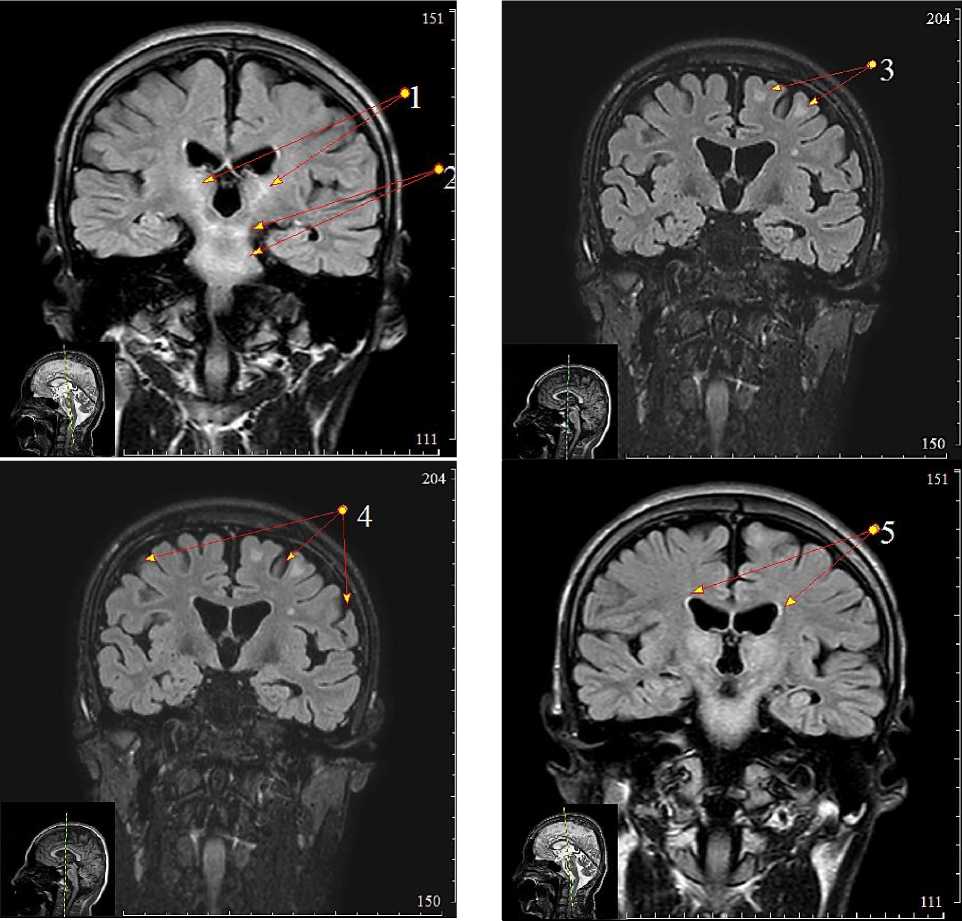
Рисунок 3. МРТ головного мозга у пациента N 28 лет с болезнью Вильсона-Коновалова в режимах FLAIR, T2, T1 и диффузно-взвешенных изображениях (DWI), в аксиальной коронарной и сагиттальной плоскостях: 1 — патологические очаги на уровне таламуса. 2 — патологические усиление интенсивности сигналов в Т2W, FLAIR режимах на уровне ножки мозга и моста. 3 — патологические очаги в лобной доле правого полушария. 4 — Расширение и углубление кортикальных борозд. 5 — перивентрикулярные мелкие очаги
В пределах белого вещества, перивентрикулярно, на уровне полу- и семиовальных центров обоих полушарий головного мозга определяются единичные мелкие очаги усиления мр-сигнала, соответствующие по сигнальным характеристикам глиоза, полициклической формы, с четкими, неровными контурами, с наибольшими размерами от 2,0 мм до 13,0 мм в поперечнике, характеризующееся с гиперинтенсивными мр-сигналами на Т2W, FLAIR и изо-гипоинтенсивными мр-сигналами на Т1W режимах исследования. В стволе мозга на уровне Варолиева моста, с частичным вовлечением задних ножек внутренней капсулы и ножек среднего мозга симметрично с обеих сторон, и распространением поражения на область подкорковых структур, определяются очаги сливающегося характера усиления мр-сигналов (соответствующие с наибольшей вероятностью к очагам глиоза сосудистого генеза, и с наименьшей — демиелинизирующим изменениям) на Т2W, FLAIR и DWI режимах исследования. Сильвиевы и кортикальные борозды умеренно выраженно неравномерно расширены и углублены.
УЗИ-органов брюшной полости от 24.01.2022 г. Печень контуры ровные, четкие; нижний край — острый. Паренхима гомогенная, диффузно н однородная, местами повышенной Заключение: Признаки хронического гепатита. Холецистит. Спленомегалия. Хронический пиелонефрит. ЭКГ от 24.01.2022 г.: Ритм синусовый. ЧСС 106 в мин. Нормальное положение ЭОС. Синусовая тахикардия. Консультации: Гастроэнтеролог от 08/02/2022 г. Диагноз: Хронический криптогенный гепатит минимальной степени активности. Спленомегалия. Окулист от 10/02/2022 г. Диагноз: Ангиопатия сетчатки OU. Синдром сухого глаза. На основании жалоб, анамнеза заболевания, возраста дебюта болезни, неврологического статуса и лабораторно-инструментальных данных выставлен диагноз: Гепатолентикулярная дегенерация (болезнь Вильсона-Коновалова), дрожательно-ригидная форма, с выраженным грубым постурально-кинетическим тремором рук и дизартрией. Хронический гепатит минимальной степени активности. Спленомегалия. В пользу диагноза свидетельствует возраст дебюта болезни, характерные жалобы со стороны нервной системы, прогрессирующее течение. В неврологическом статусе признаки поражения экстрапирамидной системы преимущественно базальных ганглиев на это указывает гиперкинез, повышение мышечного тонуса по экстрапирамидному типу и признаки поражения ствола мозга в виде нарушение функции черепно-мозговых нервов в виде дизартрии и нарушение функции слюнных желез. Кроме неврологических проявлениях при обследовании лабораторно выявлено тромбоцитопении, инструментально на УЗИ спленомегалия, МРТ головного мозга выявляется очаги перивентрикулярно, на уровне базальных ганглиев, ствола мозга и мозжечке. Из специфических лабораторных методов выявлено резкое снижение содержание меди в крови, а также церрулоплазмина, в моче отмечается повышенное выделение меди. На фоне приема пеницилламина (купренил) и цинка неврологическая симптоматика (экстрапирамидные расстройства) уменьшились. Интенсивность тремора, координаторные нарушения значительно уменьшились. Больной выписан домой с улучшением. Рекомендована постоянная хелатная терапия и препараты цинка. Дана рекомендации по диете ограничить продукты с высоким содержанием меди такие как, печень, креветки, шоколад, орехи и грибы. Исключить прием препаратов и витаминов содержащий мед.
Обсуждение. Приведенный клинический случай демонстрирует еще раз подтверждает, что классические случает в реальной клинической практике встречается не всегда; на примере нами приведенный больной в начале лечился как последствие травматического поражения головного мозга, но оказалось болезнь прогрессирует, а от полученного лечения эффекта не было. Впервые подозревает, когда уже появились выраженные экстрапирамидные расстройства в виде грубого тремора верхних конечностей, нарушения координацию в виде
Бюллетень науки и практики / Bulletin of Science and Practice Т. 9. №3. 2023 шаткости при ходьбе, затруднение самообслуживания (из-за тремора самостоятельный прием пищи была невозможной), кроме того, имелось место нарушения речи в виде дизартрии, слюнотечение, ухудшение когнитивных функций и наличие цефалического синдрома и инсомнии. В неврологическом статусе когнитивный дефицит с преимущественным нарушением кратковременной памяти, дизартрия, повышением мышечного тонуса по экстрапирамидному типу выявлено положительный симптом «Зубчатого колесо» на верхних конечностях, рефлексы орального автоматизма (+), координаторные пробы выполнял с интенцией и мимопопаданием с двух сторон, грубый, сложный тремор верхних конечностей (постурально-акционной) по типу «бьющихся крыльев». Выявляется очаги гиперинтенсивности гиперинтенсивными мр-сигналами на Т2W, FLAIR в субкортикальном белом веществе, базальных ганглиях, стволе. Базальные ганглии это один из мест, где самого начала отмечается накопления меди и соответственно в клинике отмечается экстрапирамидные расстройства. Несмотря на позднюю диагностику после начало специфической терапии состояние больного значительно улучшилась, следовательно, и качество жизни. Крайне важным является раннее распознание и начало заболевания чтобы предотвратить инвалидизируюшее последствие. Многолетняя практика показывает, что всем больным с экстрапирамидными нарушениями надо проверить наличие нарушения обмена меди особенно в молодом возрасте. Такие больные в большинстве случает вначале попадает в поле зрения семейных врачей и врачей общего профиля. Повышение осведомленности врачей первичного звенья здравоохранения о гепатолентикулярной дегенерации способствует ранней выявлению, правильной тактике диагностического поиска и лечения.
Заключение. Таким образом раннее распознавание имеет первостепенное значение, поскольку задержки в лечении имеют гораздо более высокий риск неблагоприятных клинических исходов. В данном клиническом примере диагноз болезнь Вильсона – Коновалова была выставлена поздно тогда уже появились выраженные неврологические проявления в виде выраженного экстрапирамидного расстройства в виде грубого тремора верхних конечностей. Многолетняя практика показывает, что всем больным с экстрапирамидными нарушениями надо проверить наличие нарушения обмена меди особенно в молодом возрасте. Такие больные в большинстве случает вначале попадает в поле зрения семейных врачей и врачей общего профиля. Повышение осведомленности врачей первичного звенья здравоохранения о гепатолентикулярной дегенерации способствует ранней выявлению, правильной тактике диагностического поиска и лечения.
Список литературы Болезнь Вильсона-Коновалова: обзор литературы и случай из практики
- Gerosa C., Fanni D., Congiu T., Piras M., Cau F., Moi M., Faa G. Liver pathology in Wilson's disease: From copper overload to cirrhosis // Journal of inorganic biochemistry. 2019. V. 193. P. 106-111. https://doi.org/10.1016/j.jinorgbio.2019.01.008
- Takkar B., Temkar S., Venkatesh P. Wilson disease: Copper in the eye // The National medical journal of India. 2018. V. 31. №2. P. 122-122. https://doi.org/10.4103/0970-258x.253156
- Zou L., Song Y., Zhou X., Chu J., Tang X. Regional morphometric abnormalities and clinical relevance in Wilson's disease // Movement disorders. 2019. V. 34. №4. P. 545-554. https://doi.org/10.1002/mds.27641
- Wolf T. L., Kotun J., Meador-Woodruff J. H. Plasma copper, iron, ceruloplasmin and ferroxidase activity in schizophrenia // Schizophrenia research. 2006. V. 86. №1-3. P. 167-171. https://doi.org/10.1016/j.schres.2006.05.027
- Shafritz D. A., Oertel M. Model systems and experimental conditions that lead to effective repopulation of the liver by transplanted cells // The international journal of biochemistry & cell biology. 2011. V. 43. №2. P. 198-213. https://doi.org/10.1016/j.biocel.2010.01.013
- Medici V., Czlonkowska A., Litwin T., Giulivi C. Diagnosis of Wilson disease and its phenotypes by using artificial intelligence // Biomolecules. 2021. V. 11. №8. P. 1243. https://doi.org/10.3390/biom11081243
- Lucena-Valera A., Perez-Palacios D., Muñoz-Hernandez R., Romero-Gómez M., Ampuero J. Wilson's disease: Revisiting an old friend // World journal of hepatology. 2021. V. 13. №6. P. 634. https://doi.org/10.4254%2Fwjh.v13.i6.634
- Nagral A., Sarma M. S., Matthai J., Kukkle P. L., Devarbhavi H., Sinha S., Dhawan A. Wilson's disease: clinical practice guidelines of the Indian national association for study of the liver, the Indian society of pediatric gastroenterology, hepatology and nutrition, and the movement disorders society of India // Journal of clinical and experimental hepatology. 2019. V. 9. №1. P. 74-98. https://doi.org/10.1016/j.jceh.2018.08.009
- Capone K., Azzam R. K. Wilson's Disease: A review for the general pediatrician // Pediatric annals. 2018. V. 47. №11. P. e440-e444. https://doi.org/10.3928/19382359-20181026-01
- Horn N., Møller L. B., Nurchi V. M., Aaseth J. Chelating principles in Menkes and Wilson diseases: Choosing the right compounds in the right combinations at the right time // Journal of inorganic biochemistry. 2019. V. 190. P. 98-112. https://doi.org/10.1016/j.jinorgbio.2018.10.009
- Duncan A., Yacoubian C., Beetham R., Catchpole A., Bullock D. The role of calculated non-caeruloplasmin-bound copper in Wilson’s disease // Annals of clinical biochemistry. 2017. V. 54. №6. P. 649-654. https://doi.org/10.1177/0004563216676843
- Stezin A., Kamble N., Jhunjhunwala K., Prasad S., Pal P. K. Clinical utility of longitudinal measurement of motor threshold in Wilson’s disease // Canadian Journal of Neurological Sciences. 2019. V. 46. №2. P. 251-254. https://doi.org/10.1017/cjn.2018.379
- Poujois A., Sobesky R., Meissner W. G., Brunet A. S., Broussolle E., Laurencin C., Woimant F. Liver transplantation as a rescue therapy for severe neurologic forms of Wilson disease // Neurology. 2020. V. 94. №21. P. e2189-e2202. https://doi.org/10.1212/WNL.0000000000009474
- Hamilton J. P., Koganti L., Muchenditsi A., Pendyala V. S., Huso D., Hankin J., Lutsenko S. Activation of liver X receptor/retinoid X receptor pathway ameliorates liver disease in Atp7B−/−(Wilson disease) mice // Hepatology. 2016. V. 63. №6. P. 1828-1841. https://doi.org/10.1002/hep.28406
- Ranucci G., Polishchuck R., Iorio R. Wilson’s disease: prospective developments towards new therapies // World Journal of Gastroenterology. 2017. V. 23. №30. P. 5451. https://doi.org/10.3748%2Fwjg.v23.i30.5451
- Nazer H., Ede R. J., Mowat A. P., Williams R. Wilson's disease: clinical presentation and use of prognostic index // Gut. 1986. V. 27. №11. P. 1377-1381. http://dx.doi.org/10.1136/gut.27.11.1377
- Guillaud O., Dumortier J., Sobesky R., Debray D., Wolf P., Vanlemmens C., .Duclos-Vallée J. C. Long term results of liver transplantation for Wilson’s disease: experience in France // Journal of hepatology. 2014. V. 60. №3. P. 579-589. https://doi.org/10.1016/j.jhep.2013.10.025
- Gupta S., Rajvanshi P., Lee C. D. Integration of transplanted hepatocytes into host liver plates demonstrated with dipeptidyl peptidase IV-deficient rats // Proceedings of the National Academy of Sciences. 1995. V. 92. №13. P. 5860-5864. https://doi.org/10.1073/pnas.92.13.5860
- Gupta S. Cell therapy to remove excess copper in Wilson's disease // Annals of the New York Academy of Sciences. 2014. V. 1315. №1. P. 70-80. https://doi.org/10.1111/nyas.12450
- Jaber F. L., Sharma Y., Gupta S. Demonstrating potential of cell therapy for Wilson’s disease with the Long-Evans cinnamon rat model // Hepatocyte Transplantation: Methods and Protocols. 2017. P. 161-178. https://doi.org/10.1007/978-1-4939-6506-9_11
- Murillo O., Luqui D. M., Gazquez C., Martinez-Espartosa D., Navarro-Blasco I., Monreal J. I., Gonzalez-Aseguinolaza G. Long-term metabolic correction of Wilson’s disease in a murine model by gene therapy // Journal of hepatology. 2016. V. 64. №2. P. 419-426. https://doi.org/10.1016/j.jhep.2015.09.014
- Takahashi K., Yamanaka S. A decade of transcription factor-mediated reprogramming to pluripotency // Nature reviews Molecular cell biology. 2016. V. 17. №3. P. 183-193. https://doi.org/10.1038/nrm.2016.8
- Concilli M., Iacobacci S., Chesi G., Carissimo A., Polishchuk R. A systems biology approach reveals new endoplasmic reticulum-associated targets for the correction of the ATP7B mutant causing Wilson disease // Metallomics. 2016. V. 8. №9. P. 920-930. https://doi.org/10.1039/c6mt00148c
- Członkowska A., Litwin T., Dusek P., Ferenci P., Lutsenko S., Medici V., Schilsky M. L. Wilson disease // Nature reviews Disease primers. 2018. V. 4. №1. P. 21. https://doi.org/10.1038/s41572-018-0018-3
- Roybal J. L., Endo M., Radu A., Gray L., Todorow C. A., Zoltick P. W., Flake A. W. Early gestational gene transfer with targeted ATP7B expression in the liver improves phenotype in a murine model of Wilson's disease // Gene therapy. 2012. V. 19. №11. P. 1085-1094. https://doi.org/10.1038/gt.2011.186
- Zhang S., Chen S., Li W., Guo X., Zhao P., Xu J., Esteban M. ARescue of ATP7B function in hepatocyte-like cells from Wilson's disease induced pluripotent stem cells using gene therapy or the chaperone drug curcumin // Human molecular genetics. 2011. V. 20. №16. P. 3176-3187. https://doi.org/10.1093/hmg/ddr223
- Auman J. T. Gene therapy: Have the risks associated with viral vectors been solved? // Current opinion in molecular therapeutics. 2010. V. 12. №6. P. 637-638.
- Dhawan P., Singh A. B., Deane N. G., No Y., Shiou S. R., Schmidt C., Beauchamp R. D. Claudin-1 regulates cellular transformation and metastatic behavior in colon cancer // The Journal of clinical investigation. 2005. V. 115. №7. P. 1765-1776. https://doi.org/10.1172/JCI24543
