Единый подход к созданию универсальных баз данных точных масс и времён удерживания пептидных маркеров белков на основе модели критической хроматографии биомакромолекул

Автор: Придатченко М.Л., Тарасова И.А., Массел Он К., Горшков А.В., Горшков М.В.
Журнал: Труды Московского физико-технического института @trudy-mipt
Рубрика: Оригинальные статьи
Статья в выпуске: 1 т.1, 2009 года.
Бесплатный доступ
Короткий адрес: https://sciup.org/142185573
IDR: 142185573
Текст статьи Единый подход к созданию универсальных баз данных точных масс и времён удерживания пептидных маркеров белков на основе модели критической хроматографии биомакромолекул
I. Введение I.1. Идентификация пептидов на основе «скорострельной» протеомики
В настоящее время значительная часть исследований в биологии сконцентрирована вокруг новой области — протеомики, целью которой является исследование распределения белков, их взаимодействия и модификации внутри клеток живых организмов. Задачи, решаемые протеомикой, представляют серьезное испытание для исследователей из-за разнообразия и сложности белков и пептидов, содержащихся в биологических системах. Постоянно меняющаяся природа протеома является основной сложностью его анализа: протеомные исследования требуют проведения анали- за большого количества образцов, полученных для определённых физиологических состояний или в определённых условиях окружающей среды. В свою очередь это диктует и стимулирует основную тенденцию методического развития аналитических технологий в протеомике в сторону повышения скорости и производительности анализа. Хромато-масс-спектромет-рия является одной из «базовых» аналитических технологий, используемых в про-теомных исследованиях, в которых масс-спектрометрия (МС) является основным методом, используемым в протеомике для структурной идентификации биомолекул. Одним из ключевых элементов этой технологии является разделение смесей биомолекул на входе масс-спектрометра. В большинстве протеомных исследований пептидная смесь предварительно разделяет- ся методами одномерной или двухмерной высокоэффективной жидкостной хроматографии (ВЭЖХ). Для каждого пептида измеряется молекулярный вес, а также молекулярный вес его фрагментов, полученных тем или иным методом тандемной масс-спектрометрии (МС/МС). Полученные данные о массах пептида и фрагментов затем используются в поиске по базам данных белков с целью его идентификации. Следует, однако, отметить, что только от 5 до 15% всех данных, полученных методами тандемной масс-спектрометрии, дают положительную идентификацию пептидов через базы данных белков. Причинами этого является, во-первых, проблема неточности геномных баз данных, на основе которых производится поиск и идентификации пептидов с последующим использованием полученных данных при создании баз данных ВЭЖХ-МС/МС. Во-вторых, существует проблема неэффективности и неточности методов тандемной масс-спектрометрии, на основе которых производится поиск пептидов по геномным базам данных белков с их последующей идентификацией. Фрагментация методами МС/МС может быть неполной, либо в неправильном месте, либо массы фрагментов будут измерены с недостаточной точностью (например, неправильно идентифицированы мо-ноизотопные пики в масс-спектрах фрагментов пептида). Решение этих проблем видится в интегрировании различных ортогональных методов получения информации о первичной структуре пептидов и, в первую очередь, жидкостной хроматографии. Поэтому буквально в последние несколько лет начали развиваться подходы так называемой «скорострельной» (в англоязычной литературе — shotgun) протеомики, в основе которых лежит идея использования пептидных маркеров белков. В качестве маркеров могут выступать меченные изотопами пептиды либо пептиды, уникальные для того или иного белка. В последние годы было показано, что, если масса пептида может быть измерена с точностью, достаточной для уникальной идентификации пептида, предсказанного для конкретной геномной последовательности, такой пептид может быть использован как биомаркер соответствующего родительского белка. Это в свою оче- редь даёт возможность проведения количественной высокопроизводительной идентификации белков на основе хромато-масс-спектрометрических методов анализа без использования рутинных масс-спектрометрических подходов к секвенированию, основанных на фрагментации ионов пептидов. Результатом хромато-масс-спектрометрического анализа является база данных точных масс для точно определённых пептидных последовательностей и соответствующих им хроматографических времён удерживания. Такие базы данных получили название баз данных точных масс и времён пептидных меток, или маркеров, белков (Accurate Mass and Time, AMT, tags), которые в процессе «простого» (без использования тандемной масс-спектрометрии) хромато-масс-спектрометрического анализа могут использоваться как биомаркеры на наличие в образце того или иного белка. Создание таких баз данных AMT потенциально позволит количественно идентифицировать тысячи белков за время одного анализа (порядка 1 часа) [1–4]. Несмотря на кажущуюся простоту и даже «тривиальность», развитию метода АМТ и его широкому использованию препятствует тот факт, что в отличие от масс, которые однозначно привязаны к шкале масс элементов, хроматографические данные не универсальны: они зависят от конкретных экспериментальных условий разделения и используемых инструментальных платформ. Поэтому, прежде чем включать хроматографические времена в базу данных АМТ, следует разработать процедуру приведения хроматографических времён удерживания пептидов к единой шкале и сделать их «независимыми» от условий хроматографического разделения. Решение этой задачи было основной целью работы.
-
I.2. Создание баз данных АМТ: проблема стандартизации времён удерживания
Ранее неоднократно делались попытки введения единой процедуры нормализации хроматографических данных. Например, в работе Петритиса с коллегами из Тихоокеанской северо-западной национальной лаборатории США было предложено нормализовывать экспериментальные времена удерживания приведением их к отрезку [0; 1] с помощью линейного уравнения y = к • x + m, где коэффициент m отвечает за так называемые «мёртвые» объёмы удерживания, а к — за наклон градиента [5]. При этом сами коэффициенты линейного уравнения авторы предложили находить эмпирически с помощью генетического алгоритма, широко применяющегося, в частности, в биоинформатике. В основе используемого ими генетического алгоритма лежит итерационный процесс оптимизации коэффициентов к и m [6].
Другой подход к нормализации хроматографических времён удерживания также основан на использовании линейного преобразования, коэффициенты в котором вычисляются с помощью выбранного внутреннего стандарта [7]. Причём хроматограмма реального образца делится на временные интервалы по временам выхода пептидов стандарта, и для каждого временного отрезка вычисляются коэффициенты линейного уравнения к и m на основе экспериментальных времён удерживания пептидов внутреннего стандарта. Времена удерживания пептидов, вышедших из колонки в определённом временном интервале, нормализуются в этом интервале с использованием соответствующих калибровочных коэффициентов.
Группа из исследовательского центра рака в Вашингтоне предложила использовать в качестве нормализованных времён удерживания расчётные значения гидрофобности пептидов [4], обуславливая такой подход линейной корреляцией между гидрофобностью пептидов и их хроматографическими временами удерживания [8].
Следует отметить, что применение всех предложенных до сих пор подходов к стандартизации времён удерживания ограничивается необходимостью придерживаться жёстко заданного экспериментального протокола разделения или определённого, заранее выбранного, хроматографического стандарта — калибранта. Это приводит к тому, что создаваемые таким образом хроматографические базы данных становятся привязанными к конкретным экспериментальным условиям и используемым инструментальным комплексам. Возникает серьезная дилемма: с одной стороны, создание баз данных для протеомов сложных организмов требует одновремен- ных усилий со стороны разных исследовательских групп и лабораторий в рамках совместных проектов, с другой стороны, не существует в настоящее время общего подхода к созданию баз данных, не привязанных к конкретным экспериментальным условиям. Иными словами, базы данных, особенно в части, касающейся хроматографических времён удерживания, не являются универсальными или переносимыми с одной инструментальной платформы на другую, и становится неясным, каким образом создавать базы данных, в которых заложены хроматографические времена удерживания пептидных маркеров белков. Цель настоящей работы — разработка процедуры нормализации хроматографических времён удерживания при создании баз данных АМТ, позволяющей привести экспериментальные хроматографические данные для пептидов к единой, универсальной шкале времени.
II. Результаты и обсуждение II.1. Описание экспериментальных условий
Эксперименты по разделению и идентификации индивидуальных пептидов и их смесей проводились на наносистеме Ultimate 3000 (Dionex, США), которая имела в качестве детектора масс-спектрометр на основе линейной радиочастотной ионной ловушки LTQ (Thermo Fisher, Бремен, Германия). Масс-спектрометр был оборудован наноэлектроспреем (Proxeon Biosystems, Оденсе, Дания), позволяющим получать многозарядные макромолекулярные ионы, которые затем фрагментировались в ионной ловушке в столкновениях с молекулами буферного газа (CAD). Разделение смесей пептидов производилось при скоростях подачи растворителя в диапазоне от 200 до 400 нл / мин для различных профилей градиента бинарной смеси растворителя вода-ацетонит-рил в диапазоне от 0 , 3% B до 1 , 7% B в минуту. Время нарастания градиента варьировалось от 30 до 120 мин. Мобильная фаза состояла из двух компонентов: (А) 98% воды, 1 , 9% ацетонитрила, 0 , 1% муравьиной кислоты и (В) 80% ацетонитрила, 19 , 9% воды, 0 , 1% муравьиной кислоты.
В работе использовались коммерческие стандарты протеолитических смесей белков: дайджест белка Cytochrome C и дайджест шести белков 6 Protein Mixture, производимые фирмой Dionex/LCPacking (USA).
Для расчётов теоретических времён удерживания нами была использована модель предсказания хроматографических объёмов/времён удерживания биомолекул по их аминокислотной последовательности на основе концепции жидкостной хроматографии в критических условиях [11], реализованная в виде программного обеспечения «Теоретический хроматограф». «Теоретический хроматограф» позволяет предсказывать времена удерживания белков и пептидов в зависимости от их первичной структуры при заданных экспериментальных параметрах разделения: (1) параметры колонки (внутренний диаметр и длина колонки, размер пор, фактор, показывающий долю пространства в колонке, занятую твёрдой фазой); (2) профиль градиента и (3) состав компонентов растворителя. Для расчёта времён удерживания «Теоретический хроматограф» использовались полученные нами ранее [11, 12] эффективные энергии адсорбции для 20 наиболее часто встречающихся природных аминокислот, а также C- и N-концевых групп.
-
II.2. Концепция линейной корреляции хроматографических времён удерживания
В основе подхода, рассмотренного в данной работе, лежит предположение о линейной корреляции времён удерживания пептидов, полученных в разных условиях разделения, наиболее широко используемых в протеомных исследованиях. Действительно, если у нас наблюдается линейная корреляция между хроматографическими данными, полученными в различных условиях разделения, то достаточно выбрать определённые условия в качестве стандарта и «привязывать» экспериментальные данные к данным, соответствующим этим стандартным условиям через элементарную процедуру нормировки. Нормализованные таким образом времена удерживания можно вносить в базы данных, поскольку в этом случае пептид А в любых экспериментах будет иметь после нормализации те же абсолютные значения точных времен, что и в базе данных.
-
II.3. Экспериментальная проверка концепции линейности хроматографических данных
Для проверки, насколько применима концепция линейной корреляции экспериментальных хроматографических времён удерживания и определения диапазона экспериментальных параметров разделения, в рамках которых она выполняется, нами был проведён ряд экспериментов по выявлению влияния на времена удерживания следующих параметров разделения: скорость градиента, скорость потока бинарной смеси растворителя, характеристики колонок. Параметры использованных хроматографических колонок и их производители представлены в табл. 1. Влияние указанных параметров также было исследовано с использованием различных хроматографических систем от разных производителей.
Таблица 1
Параметры хроматографических колонок, использовавшихся в экспериментах
|
Колонка |
Производитель |
Фаза |
i.d., мкм |
Длина, мм |
Размер частиц, мкм |
Размер пор, ˚A |
|
1 |
LC-Parcking |
PepMap C18 |
75 |
150 |
3 |
100 |
|
2 |
LC-Parcking |
PepMap C18 |
75 |
250 |
3 |
100 |
|
3 |
Waters |
Atlanties C18 |
75 |
150 |
3 |
100 |
|
4 |
LC-Parcking |
PepMap C18 |
75 |
150 |
5 |
100 |
|
5 |
LC-Parcking |
PLRP-S |
75 |
150 |
5 |
300 |
|
6 |
Merk |
Si Monolith |
100 |
150 |
N/A |
130 |
Таблица 2
Коэффициенты линейной корреляции экспериментальных времён удерживания пептидов протеолитической смеси пептидов белка Cytochrome C для различных условий разделения, использовавшихся колонок и протоколов
|
Наклон градиента |
||||||
|
1 , 7% В/ мин |
1 , 2% В/ мин |
1 , 4% В/ мин |
0 , 8% В/ мин |
0 , 4% В/ мин |
0 , 3% В/ мин |
|
|
Колонка 1 |
0 , 992 |
0 , 998 |
0 , 998 |
0 , 997 |
0 , 995 |
0 , 988 |
|
Скорость потока растворителя |
||||||
|
200 нл / мин |
300 нл / мин |
400 нл / мин |
||||
|
0 , 8% В / мин |
0 , 998 |
0 , 998 |
0 , 997 |
|||
|
Колонка |
||||||
|
1 |
2 |
3 |
4 |
5 |
6 |
|
|
1 , 7% В / мин |
0 , 994 |
0 , 994 |
0 , 978 |
0 , 992 |
0 , 974 |
0 , 990 |
|
0 , 3% В / мин |
0 , 988 |
0 , 992 |
0 , 966 |
0 , 990 |
0,967 |
0 , 988 |
-
• Пептиды дайджеста белка Cytochrome с
-
• Пептиды дайджеста 6 белков стандарта 120 1
| 20
£
0О 20 40 60 80 100 120
RTexp, мин (ВЭЖХ система 1)
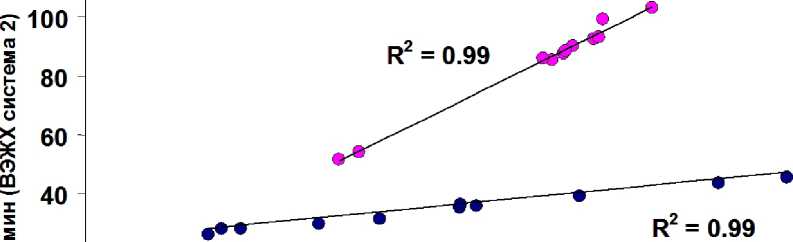
Рис. 1. Линейная корреляция времён удерживания пептидов протеолитического дайджеста белка Cytochrome C и коммерческого стандарта, состоящего из 6 белков, полученных в различных лабораториях для различных экспериментальных условий и протоколов разделения. Система 1: градиентный ВЭЖХ хроматограф Agilent 1200 с колонкой на обращенной фазе (ИНЭП ХФ РАН, Москва). Система 2: градиентный ВЭЖХ хроматограф Dionex Utlimate 3000 с колонкой на обращенной фазе (CEA, Гренобль). Для обеих систем использовались различные протоколы разделения, отличающиеся как профилем градиента, так и составом бинарной смеси растворителей
На рис. 1 представлен пример корреляции между хроматографическими временами удерживания, полученных в разных лабораториях для протеолитических смесей пептидов белка Cytochrome C и стандартного набора 6-ти белков. В табл. 2 суммированы результаты измеренных коэффициентов линейной корреляции для различных протоколов и параметров разделения. Как видно из представленной таблицы, наблюдается высокая линейная корреляция между экспериментальными данными, что подтверждает изначальное предположение о «линейности» хроматографических данных. Следует отметить, что выбранные нами протоколы и параметры разделения охватывают практически весь диапазон экспериментальных параметров, используемых в современных протеомных исследованиях при решении задачи идентификации и количественного анализа белков на основе хромато-масс-спектрометрии и протеомных баз данных. Осталось только понять, всегда ли при- менима концепция «линейности» хроматографических данных для пептидов. На первый взгляд, концепция линейности выглядит тривиальной. Действительно, если обратиться к базовому уравнению жидкостной хроматографии:
Vyдерживание VMёртвый объём + KD ' Vобъём пор, где KD — хроматографический коэффициент распределения, то, поскольку KD пропорционален энергии адсорбции и не зависит от условий разделения (для одной и той же кислотности и ряда других химических свойств бинарной смеси растворителя), линейность между временами удерживания должна всегда наблюдаться. Однако это упрощённый взгляд на природу разделения пептидов, который исхо- дит из представления о чисто адсорбционном механизме разделения по аналогии с разделением низкомолекулярных соединений. Вместе с тем ранее нами было показано [11–13], что разделение пептидов нельзя рассматривать по аналогии с разделением низкомолекулярных соединений, поскольку для них, как и в случае макромолекул, существуют три режима разделения в зависимости от хроматографических условий: адсорбционный (разделение идёт через механизмы адсорбции), эксклюзионный (определяемый энтропийными факторами) и переходный, критический (между эксклюзией и адсорбцией). В зависимости от режима разделения в случае пептидов наблюдается изменение порядка выхода из хроматографической колонки.
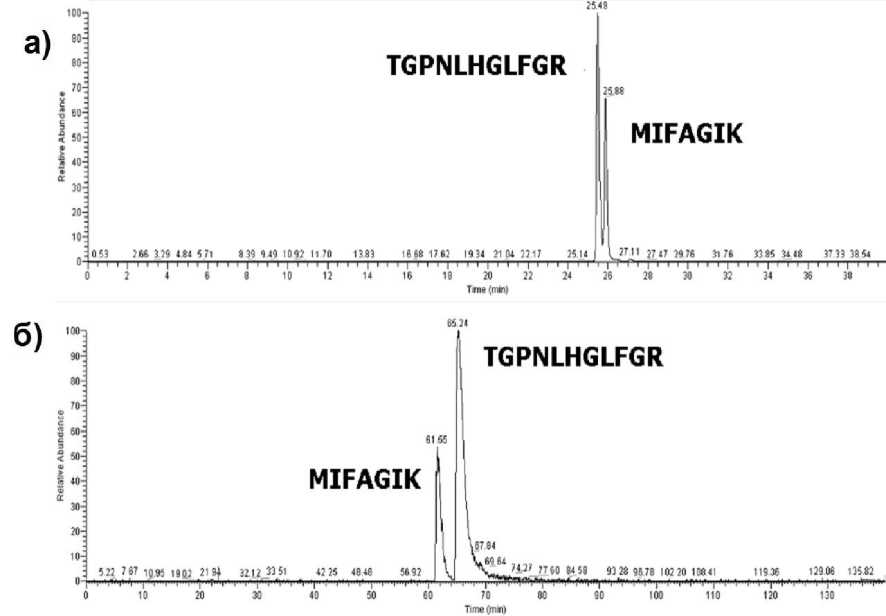
Рис. 2. Изменение порядка выхода пептидов, имеющих близкие времена удерживания, при изменении условий градиентного элюирования. Хроматограммы пептидов TGPNLHGLFGR и MIFAGIK, полученные при изменении наклона градиента с 1 , 7% В/ мин (2а) на 0 , 3% В/ мин (2б)
На рис. 2 приведён пример, полученный нами для протеолитической смеси пептидов белка Cytochrome C, который иллюстрирует обозначенную выше проблему. Пептиды TGPNLHGLFGR и MIFAGIK из этой смеси имеют близкие значения времён удерживания. При изменении скорости градиента меняется порядок выхода этих пептидов из хроматографической ко- лонки. Это наглядный пример проявления «макромолекулярной» сущности поведения биомолекул в условиях хроматографического разделения на обращенной фазе. При быстром градиенте разделение пептидов идёт в эксклюзионном режиме и время удерживания определяется не адсорбцией пептидов (как в случае медленного градиента), а длиной аминокислотной последовательности пептидов. В результате более длинный пептид TGPNLHGLFGR выходит раньше, несмотря на то, что его адсорбционные свойства выше, чем у более короткого пептида MIFAGIK. Таким образом, проблема линейности между экспериментальными хроматографическими временами удерживания для пептидов не является тривиальной и требует дальнейших систематических исследований. На основе полученных данных мы можем только констатировать тот факт, что в широком диапазоне и, что особенно важно, в пределах экспериментальной ошибки измерения времён удерживания пептидов в современной хроматографии концепция «линейности» соблюдается. Это позволяет утверждать о возможности введения единой (с точки зрения протоколов разделения или используемых хроматографических систем) шкалы времён удерживания, или, точнее, нормализованных времён удерживания, на основе простых линейных соотношений. Таким образом, задача сводится к нахождению процедуры такой нормализации, которая позволит избежать ограничений чисто эмпирических подходов, упомянутых выше, и при этом решить проблемы, связанные с изменениями порядка выхода пептидов при изменении экспериментальных параметров, пример которых представлен на рис. 2.
-
II.4. Модель BioLCCC как основа единой шкалы времён удерживания
Для решения проблемы нормализации экспериментальных данных при создании баз данных пептидных маркеров белков нами предлагается использовать методы предсказания теоретических времён удерживания в зависимости от аминокислотных последовательностей пептидов и конкретных условий разделения. В основе предлагаемого подхода лежит ранее предложенная нами модель хроматографического разделения пептидов BioLCCC, развитая на основе критической хроматографии биомакромолекул [11–13]. Модель BioLCCC позволяет однозначно предсказывать времена удерживания пептидов в зависимости от их аминокислотной последовательности, наличия и локализации посттрансляционных модификаций, а также в комбинации с масс-спектрометрией
ТРУДЫ МФТИ, 2009, Том 1, № 1 высокого разрешения определение неизвестных последовательностей (секвенирование de novo) [14]. Используя теоретическое предсказание времён удерживания для выбранного пептидного стандарта (например, протеолитическая смесь пептидов белка Cytochrome C), можно ввести новую шкалу времени, в которой экспериментальные значения времён удерживания исследуемых пептидов будут иметь одни и те же (для одних и тех же пептидов) нормализованные времена. В основе предлагаемой процедуры нормализации лежит рассмотренное выше предположение о том, что хроматографические данные, полученные для одних и тех же образцов на различных хроматографических системах с использованием различных хроматографических колонок, состава мобильной фазы и профиля градиента, линейно коррелируют, то есть для одного и того же пептида
RT эксп.
1 — a • RT эксп . 2 + b.
Дальнейшее построение шкалы нормализованных времён делается следующим образом: выбирается пептидный стандарт, для которого рассчитываются теоретические значения времён удерживания в условиях единого, заранее определённого протокола разделения («базисный» протокол). Далее теоретические времена удерживания для пептидов стандарта нормализуются в диапазоне [0; 1] , где время удерживания для наиболее гидрофобного пептида стандарта выбирается равным 1 , 0 (в принципе приведение шкалы времени в отрезок [0; 1] необязательно, однако представляется более удобным с точки зрения общности шкалы). Подставляя в линейное уравнение экспериментальные и теоретические значения времён удерживания RT st,i для пептидов стандарта, определяются калибровочные коэффициенты:
RT st,i (теор . норм . )
a • RT st,i (эксп . ) + b.
Используя полученные калибровочные коэффициенты a и b , все экспериментальные времена RT exp,i для исследуемых пептидов переводятся в новую шкалу нормализованных времён с помощью опять же линейного уравнения
RT exp,i (норм . ) a • RT exp,i (эксп . ) + b-
Рассмотренная процедура нормализации схематически представлена на рис. 3.
Ясно, что поскольку шкала времени привязана к теоретическим временам удерживания стандарта в одних и тех же условиях разделения, то после нормализации экспериментальные времена удерживания пептидов, полученные в различных условиях разделения, при использовании различных профилей градиентов и разных хроматографических систем, становятся едиными для всех пептидов. Предложен- ная процедура аналогична процедуре калибровки в масс-спектрометрии, при которой измеряемые физические величины (например, циклотронные частоты в масс-спектрометрии ионного циклотронного резонанса, или времена пролёта во время-пролётных масс-спектрометрах) привязываются к единой шкале масс на основе углерода 12C.
Таблица 3
Сравнение экспериментальных времён удерживания пептидов TGPNLHGLFGR и MIFAGIK с предсказанными на основе разработанной авторами модели BioLCCC для различных профилей градиента
|
Градиент от 0 до 50% B вте-чение 30 минут |
Градиент от 0 до 35% B вте-чение 120 минут |
|||
|
Пептид |
TGPNLHGLFGR |
MIFAGIK |
TGPNLHGLFGR |
MIFAGIK |
|
RTexp, мин |
20,31 |
20,64 |
64,89 |
61,45 |
|
RTтеор, мин |
20,37 |
21,31 |
64,8 |
60,45 |
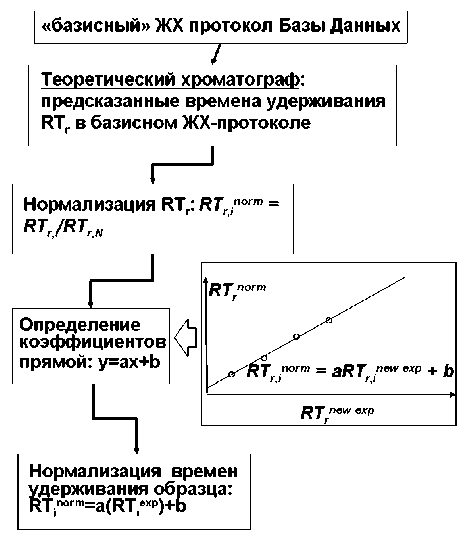
Рис. 3. Схематическое представление предложенной в работе процедуры нормализации экспериментальных хроматографических данных для пептидов на основе модели BioLCCC
Очевидно, что для предложенной процедуры нормализации пептиды, меняющие свой порядок выхода из колонки, представляют определённую сложность: никаким линейным преобразованием времён удерживания мы не получим инверсии порядка выхода пептидов при изме- нении протокола элюирования. Использование модели BioLCCC для предсказания времён удерживания позволяет решить эту проблему. Действительно, поскольку модель BioLCCC основана на «макромолекулярном» представлении о процессе разделения пептидов в условиях градиентного элюирования, эта модель является на сегодняшний день единственной, учитывающей переход от адсорбционного к эксклюзионному режиму разделения для пептидов. В табл. 3 представлены результаты расчёта теоретических времён удерживания для пептидов TGPNLHGLFGR и MIFAGIK из рассмотренного выше примера инверсии выхода при изменении скорости нарастания градиента (рис. 2). Как видно из этой таблицы, модель BioLCCC не только правильно предсказывает порядок выхода указанных пептидов, но и даёт правильные количественные оценки изменения абсолютных значений времён выхода для конкретных хроматографических условий. Для решения проблемы инверсии в задаче нормализации времён удерживания можно предложить следующий подход: с помощью модели BioLCCC проверяется наличие инверсии порядка выхода близких по удерживанию пептидов при переходе от реального хроматографического протокола к «базисному», в котором создаётся единая шкала нормализованных времен. В случае обнаружения такой инверсии данным пептидам приписываются теоретические нормализованные времена удерживания, которые и используются в дальнейшем при создании баз хроматографических данных.
-
II.5. Экспериментальная проверка нормализации времён удерживания пептидов на основе модели BioLCCC
Экспериментальная проверка предложенной процедуры нормализации осуществлялась на примере протеолитической смеси белка Cytochrome C, который нами был выбран в качестве стандарта. В этом смысле полученные данные можно рассматривать как аналогию определения точности калибровки масс-спектрометров на основе сравнения экспериментальных и теоретических масс веществ калибранта после калибровки. В табл. 4 представлены результаты обработки и нормализации времён удерживания пептидов дайджеста, полученные в результате измерений, проведённых при использовании различных хроматографических систем и протоколов разделения. В качестве «базисного» протокола нами был выбран так называемый протокол Pilot, предлагаемый фирмой Dionex/LCPacking: 60 мин градиент со скоростью нарастания градиента 0,8%В/мин, в бинарной смеси вода ацетонитрил (фаза А 98% вода /2% ацетонитрил; фаза В 20% вода /80% ацетонитрил) с кислотностью растворителя pH =2, пропускаемой через капиллярную наноколонку (75 мкм внутренний диаметр, 150 мм длина) на обращенной фазе C18 со скоростью потока 300 нл/мин. Полученные предварительные данные свидетельствуют о том, что предложенная процедура нормализации хроматографических времён удерживания пептидов позволяет получать универсальные времена удерживания с точностью порядка 1–2%, что сравнимо с точностью хроматографических экспериментов.
Таблица 4
АМТ базы данных для протеолитической смеси белка Cytochrome C
|
№ |
Последовательность |
M |
m/z |
(I) |
(II) |
(III) |
Ср. зн. |
|
1 |
KYIPGTK |
805,5 |
403,8 |
0,3532 |
0,3540 |
0,3692 |
0,3588 |
|
2 |
YIPGTK |
677,4 |
678,4 |
0,3883 |
0,3853 |
0,3804 |
0,3847 |
|
3 |
IFVQK |
633,4 |
634,4 |
0,4199 |
0,4176 |
0,4010 |
0,4128 |
|
4 |
KTGQAPGFSYTDANK |
1583,8 |
528,9 |
0,4821 |
0,4949 |
0,5053 |
0,4941 |
|
5 |
TGQAPGFSYTDANK |
1455,7 |
729 |
0,5520 |
0,5469 |
0,5611 |
0,5533 |
|
6 |
KGEREDLIAYLK |
1433,8 |
479,1 |
0,6382 |
0,6399 |
0,6708 |
0,6496 |
|
7 |
TGPNLHGLFGR |
1167,6 |
390,4 |
0,6694 |
0,6767 |
0,6557 |
0,6673 |
|
8 |
MIFAGIK |
778,4 |
390,3 |
0,6850 |
0,6859 |
0,6562 |
0,6757 |
|
9 |
EDLIAYLK |
963,5 |
482,9 |
0,7989 |
0,7817 |
0,7692 |
0,7833 |
|
10 |
IFVQKCAQCHTVEK |
1632,8 |
545,5 |
0,8408 |
0,8439 |
0,8861 |
0,8569 |
|
11 |
GITWGEETLMEYLENPKK |
2137 |
713,7 |
0,9326 |
0,9464 |
0,9581 |
0,9457 |
|
12 |
GITWGEETLMEYLENPK |
2008,9 |
1005,7 |
1,0185 |
1,0079 |
1,0131 |
1,0132 |
Полученные нами времена удерживания нормализованы по предложенной в работе процедуре, основанной на использовании модели критической хроматографии биомакромолекул BioLCCC. Нормализованные времена удерживания представлены в колонках (I) — (III), соответствующих различным условиям градиентного элюирования: (I) 4 – 50% B в течение 60 мин, (II) 0 – 50% B в течение 30 мин, (III) 0 – 35% B в течение 120 мин. Средняя ошибка нормализованных времён (отклонение нормализованного времени от среднего для каждого из пептидов), полученных для различных экспериментальных условий, составляет около 1%
III. Благодарности
Работа была выполнена при финансовой поддержке Российского фонда фундаментальных исследований (РФФИ, грант 08-04-01339 и 08-04-91121-АФГИР), Международной ассоциации (INTAS, грант Genomics 05-10000004-7759) и Отделения химических наук и материалов РАН (программа ОХНМ 4.2 «Создание эффективных методов химического анализа и исследования структуры веществ и материалов»).
