Генетические причины аритмического фенотипа некомпактной кардиомиопатии

Автор: Комиссарова С.М., Чакова Н.Н., Ринейская Н.М., Долматович Т.В., Ниязова С.С.
Журнал: Евразийский кардиологический журнал @eurasian-cardiology-journal
Рубрика: Оригинальные статьи
Статья в выпуске: 2, 2021 года.
Бесплатный доступ
Цель работы. Оценить генотип-фенотип ассоциации у белорусских пациентов с некомпактной кардиомиопатией (НКМ) и клинически значимыми желудочковыми аритмиями.Материалы и методы. В исследование включены 170 неродственных пациентов с НКМ, проспективно наблюдаемых в РНПЦ «Кардиология» в течение 36 мес. [6; 42,0], у которых были данные 24 - часового холтеровского мониторирования ЭКГ в течение 12 месяцев после вступления в исследование. Медиана возраста пациентов, вступивших в исследование, составляла 42 [18;69] года, преобладали мужчины (63,2%). Аритмический фенотип НКМ был диагностирован по наличию необъяснимых синкопальных состояний; неустойчивой желудочковой тахикардии (ЖТ), ≥ 500 желудочковых экстрасистол за сутки. Диагноз НКМ устанавливали на основании эхо кардиографических (ЭхоКГ) критериев Jenni и критериев магнитно-резонансной томографии (МРТ) S. Petersen и А. Jaquier. Поиск мутаций в кодирующих последовательностях 174 генов проведен 30 неродственным пациентам с НКМ методом высокопроизводительного секвенирования (NGS).Результаты. У 76 из 170 (44,7%) пациентов клинически значимые аритмии являлись ведущим проявлением заболевания. Неустойчивая ЖТ была зафиксирована у 54 (71,1%) пациентов, устойчивая ЖТ - у 11 (14,5%) пациентов, ЖЭ более 500 в сутки - у 50 (65,8%). За период наблюдения (медиана наблюдения 36 месяцев) имплантируемые кардиовертеры-дефибрилляторы (ИКД) и ресинхронизирующие устройства с функцией дефибриллятора (CRT-D) были установлены 16 (21,1%) пациентам. В результате проведения NGS у 26 (86,7%) пробандов с аритмическим фенотипом НКМ обнаружено 40 изменения нуклеотидной последовательности (5 патогенных мутаций, 30 вариантов с неопределенной значимостью (VUS), 5 новых замен) в 27 генах. Доля мутаций в генах саркомерных протеинов составила 26,9%, а в генах белков ионных каналов оказалась равной 23,1%. На долю нуклеотидных изменений в генах, кодирующих структурные белки, пришлось 11,5%. В 38,5% случаев выявлена не одна, а две или более редких мутаций, причем в 30,8% аминокислотные изменения затрагивали белки разных функциональных классов.Выводы. В группе пациентов с аритмогенным фенотипом НКМ доля лиц с наличием мутаций в генах, ассоциированных с различными кардиомиопатиями, составила 86,7% и была существенно выше, чем сообщалось у пациентов с НМП в целом (59%). Более высокой оказалась и частота встречаемости множественных мутаций (38,5%) в этой когорте. Генетические особенности пациентов наряду с клиническими характеристиками являются маркерами высокого риска развития жизнеугрожающих аритмий и могут дополнительно использоваться для прогнозирования неблагоприятных событий у пациентов с НКМ, а также для ранней диагностики заболевания у их ближайших родственников.
Некомпактная кардиомиопатия, аритмический фенотип, жизнеугрожающие аритмии, высокопроизводительное секвенирование (ngs)
Короткий адрес: https://sciup.org/143176201
IDR: 143176201 | DOI: 10.38109/2225-1685-2021-2-62-69
Arrhythmic phenotype of non-compaction cardiomyopathy
Purpose. To evaluate the genotype-phenotype association in Belarusian patients with non-compaction cardiomyopathy (NCCM) and clinically significant ventricular arrhythmias.Materials and methods. The study included 170 unrelated patients with NCCM prospectively observed in the RSPC “Cardiology” for 36 months. [6; 42,0], who underwent 24-hour Holter ECG monitoring for 12 months after entering the study. The median age of patients was 42 [18; 69] years, men - 63,2%. The arrhythmic phenotype of NCСM was diagnosed by the presence of unexplained syncope; nonsustained ventricular tachycardia, the presence of ≥500 ventricular premature beats (VPB) per day. The diagnosis of NCCM was established on the basis of the following criteria: 1) Echocardiography of the Jenny criteria; 2) CMR of the S. Petersen and A. Jaquier criteria. The mutations search in the coding sequences of 174 genes was performed in 30 unrelated patients with NCCM using next generation sequencing (NGS).Results. In 76 out of 170 (44,7%) patients, clinically significant arrhythmias were the leading manifestation of the disease. Nonsustained VT was recorded in 54 (71,1%) patients, sustained VT - in 11 (14,5%) patients, VPB more than 500 per day - in 50 (65,8%). During the follow-up period (median follow-up of 36 months), devices (ICD/CRT-D) were implanted in 16 (21,1%). NGS sequencing revealed 40 changes in the nucleotide sequence (5 pathogenic mutations, 30 variants with uncertain significance (VUS), 5 new substitutions) in 27 genes in 26 (86,7%) probands with the arrhythmic phenotype NCCM. The proportion of mutations in sarcomeric proteins genes of was 26,9%, and in ion channel proteins genes was 23,1%. Nucleotide changes in genes encoding structural proteins accounted for 11,5%. In 38,5% of cases, not one, but two or more rare mutations were detected, and in 30,8%, amino acid changes affected proteins of different functional classes.Conclusions. In the group of patients with the arrhythmic NCCM phenotype, the proportion of individuals with genes mutations associated with various cardiomyopathies was 86,7% and was significantly higher than reported in patients with NCCM generally (59%). The frequency of multiple mutations was also higher (38,5%) in this cohort. The genetic characteristics of patients, along with their clinical characteristics, are markers of a high risk of developing life-threatening arrhythmias and can be additionally used for predicting adverse events in patients with NCCM, as well as for early diagnosis of the disease in their relatives.
Текст научной статьи Генетические причины аритмического фенотипа некомпактной кардиомиопатии
Некомпактная кардиомиопатия (НКМ) – клинически и генетически гетерогенная первичная кардиомиопатия, которая характеризуется интенсивно развитыми желудочковыми трабекулами в сочетании с глубокими выстланными эндокардом межтрабекулярными лакунами, не связанными с коронарным кровотоком и предрасполагающими к образованию тромбов [1]. Результаты популяционных исследований, проведенных в различных странах, свидетельствуют о том, что распространенность НКМ в общей популяции составляет 1:5000 человек, а среди взрослых пациентов с сердечной недостаточностью (СН) – от 3% до 4% [2]. Клинические проявления заболевания разнообразны, НКМ встречается как в изолированной форме, так и в сочетании с другими кардиомиопатиями и врожденными пороками сердца. Одной из основных причин НКМ являются мутации в генах, ассоциируемых с развитием различных кардиомиопатий и кодирующих саркомерные, структурные и регуляторные белки, а также белки, ответственные за функционирование ионных каналов [3]. Мутации наследуются по аутосомно-доминантному типу, реже являются рецессивными или X-сцепленными [4].
Высокая частота жизнеугрожающих желудочковых аритмий с повышенным риском внезапной сердечной смерти (ВСС) у пациентов с НКМ, отмечалась во многих исследованиях [5,6]. В мета-анализе, проведенном Kayvanpour E. с соавт. [7], по данным 35 исследований, включивших 7598 пациентов с НКМ, было показано, что примерно у одной пятой пациентов с НКМ были выявлены жизнеугрожающие аритмии. Однако на сегодняшний день в рекомендациях Европейского общества кардиологов нет каких-либо конкретных критериев выделения пациентов высокого риска развития жизнеугрожающих желудочковых аритмических событий среди пациентов с НКМ, и показания для имплантации кардиовертера-дефибриллятора (ИКД) следуют общему руководству стратегии первичной и вторичной профилактики ВСС c использованием критериев для дилатационной кардиомиопатии (ДКМП) [8]. В 2019 г. американскими экспертами разработан консенсусный документ в отношении НКМ, в котором освещается подход к имплантации КД у этой категории пациентов [9]. Указано, что пациентам с НКМ и желудочковыми тахиаритмиями, ассоциированными с синкопальными состояниями, или ВСС с успешными реанимационными мероприятиями при ожидаемой выживаемости более 1 года, рекомендована терапия КД с целью вторичной профилактики ВСС. Имплантация КД также целесообразна у пациентов с НКМ и доказанной неустойчивой ЖТ при наличии снижения систолической функции миокарда с целью первичной профилактики ВСС [10].
Критерии стратификации риска пациентов с НКМ, разработанные к настоящему времени, были сосредоточены, в основном, на клинико-инструментальных характеристиках, включая семейный анамнез, электрокардиографические(ЭКГ) характеристики, параметры ЭхоКГ-исследования, наличии зон фиброза миокарда по данным МРТ с отсроченным контрастированием без учета генетических факторов. Однако анализ данных многочисленных исследований указывает на существование ассоциаций генотипа и фенотипа заболевания и связи неблагоприятных клинических событий с различными генотипами [11].
Аритмические генотипы, связанные с повышенным риском ВСС, хорошо известны при кардиомиопатиях, таких как ДКМП, аритмогенная кардиомиопатия правого желудочка (АКПЖ), а также гипертрофическая кардиомиопатия (ГКМП) [12]. Генетическая составляющая также играет важную роль в патогенезе НКМ. В ряде крупных исследований изучался спектр генетических причин развития злокачественных желудочковых тахиаритмий при этой патологии. В обзоре J.I. van Waning [13] при изучении результатов 172 исследований, включивших 561 пациентов с НКМ, выделены группы генов, ассоциированные с аритмиями, куда вошли гены, кодирующие белки ионных каналов: ABCC9, ANK2, CACNА2D1, CASQ2, HCN4, KCNE3, KCNH2, KCNQ1, RYR2 и SCN5А, а также генетические варианты в саркомерных и несаркомерных генах, которые часто ассоциируются с желудочковыми тахиаритмиями и имеют риск неблагоприятного исхода: TTN, RBM20, DSP, DTNA, JUP, LMNA, PKP2, PLN, PRDM16 и SGCD.
Цель работы. Оценить генотип-фенотип ассоциации у белорусских пациентов с НКМ и клинически значимыми желудочковыми аритмиями.
МАТЕРИАЛЫ И МЕТОДЫ
В исследование включены 170 неродственных пациентов с НКМ проспективно наблюдаемых в РНПЦ «Кардиология» в течение 36 мес. [6; 42,0], у которых были данные 24 – часового холтеровского мониторирования ЭКГ в течение 12 месяцев после вступления в исследование. Все участники дали добровольное письменное информированное согласие на участие в исследовании и использование соответствующих биоматериалов. Аритмический фенотип НКМ был диагностирован по наличию одного из следующих признаков: 1) необъяснимые синкопальные состояния (вероятно, из-за ЖТ); 2) неустойчивая ЖТ, определяемая как ≥ 3 последовательных желудочковых сокращений продолжительностью <30 сек с частотой ≥ 120 уд/ мин; 3) ≥ 500 желудочковых экстрасистол за сутки и был выявлен у 76 (44,7%) пациентов, медиана возраста которых составляла 42 года (18; 69); женщин – 28 (36,8), мужчин – 48 (63,2%). Клинико-инструментальное обследование помимо стандартных методов обследования (осмотр, сбор индивидуального и семейного анамнеза, ЭКГ, суточное мониторирование ЭКГ по Холтеру, ЭхоКГ) включало МРТ с отсроченным контрастированием, при необходимости коронароангиографическое исследование. Диагноз НКМ устанавливали на основании следующих критериев: 1) ЭхоКГ критериев Jenni и соавт. [14], включающих наличие соотношения некомпактного (NC) и компактного (C) слоев NC/C >2,0 в конце систолы; многочисленных чрезмерно выдающихся трабекул и глубоких межтрабекулярных углублений; наличие более 2-х трабекулярных углублений, снабжаемых внутрижелудочковой кровью по данным цветного допплеровского анализа; 2) МРТ-критериев (Petersen) при конечно-диастолическом соотношении NC/C ≥ 2,3 в одном из сегментов ЛЖ по длинным осям МРТ-изображения [15] и доли некомпактного миокарда >20% согласно критериям А. Jaquier [16].
Протокол МРТ- сканирования включал градиент-эхо последовательности с яркой кровью в кино-режиме для морфологической и функциональной оценки, градиент-эхо последовательности инверсия-восстановление с отсроченным контрстированием через 10 мин после введения контрастного вещества (омнискан) в расчете 0.1 ммоль/кг. Оценка локализации и характера отсроченного распределения контрастного препарата проводилась в 17 сегментах миокарда левого желудочка на срезах по короткой оси ЛЖ на базальном уровне (6 сегментов), среднем уровне (на уровне папиллярных мышц, 6 сегментов), на верхушечном уровне (5 сегментов). Всем пациентам, выполнившим МРТ сердца, визуально проводился анализ участков миокарда с отсроченным накоплением контрастного вещества. Для того, чтобы количественно оценить контрастное накопление препарата на каждом срезе по короткой оси ЛЖ в каждом сегменте вручную обводилась и полуавтоматически рассчитывалась площадь участка с 48 отсроченным контрастированием, затем все полученные результаты суммировались. Объем контрастированного миокарда определялся с помощью умножения суммированной площади с накоплением контраста на толщину среза.
Генетическое исследование. Поиск мутаций в кодирующих последовательностях 174 генов, ассоциируемых с сердечно-сосудистой патологией, проведен 30 неродственным пациентам с НКМ методом высокопроизводительного секвенирования (NGS) на генетическом анализаторе MiSeq (Illumina) с использованием набора «TruSight™ Cardio Sequencing Panel». Обработка и аннотирование результатов секвенирования проводилась с помощью программного обеспечения ANNOVAR rev. 527 [16], позволяющего оценить патогенность выявленного генетического варианта.
Статистическая обработка материала проводилась с использованием пакета прикладных программ «Statistica for Windows 12». Данные в таблицах представлены в виде среднего арифметического значения и стандартного отклонения, а также в виде значений медианы и 25- и 75-го процентилей (интерквартильный размах). Статистически значимыми считали различия при p <0,05.
Результаты и обсуждение
Исходная клиническая характеристика пациентов с клинически значимыми желудочковыми аритмиями представлена в таблице 1.
У 76 из 170 (44,7%) пациентов с самого начала манифестации заболевания клинически значимые желудочковые аритмии выходили на первый план и являлись ведущим проявлением заболевания. Медиана возраста пациентов, вступивших в исследование, составляла 42 [18; 69] года, преобладали мужчины (63,2%). Неустойчивая ЖТ была наиболее частым видом аритмии и зафиксирована у 54 (71,1%) пациентов, устойчивая ЖТ – у 11 (14,5%) пациентов, ЖЭ более 500 в сутки – у 50 (65,8%), хроническая форма ФП с эпизодами неустойчивой ЖТ отмечена у 34 (44,7%) пациентов. Симптомы ХСН имелись у всех пациентов, ХСН III ФК NYHA отмечена у 20 (26,3%) пациентов. У 35 (46%) наблюдалась систолическая дисфункция ЛЖ, медиана ФВ ЛЖ составила 41% [17; 80]. По данным ЭхоКГ-исследования у большинства обследуемых регистрировали дилатацию ЛЖ, так как в данной когорте преобладали пациенты с дилатационным фенотипом НКМ (53,9%). По данным МРТ с отсроченным контрастированием у 37 (48,7%) пациентов обнаружены зоны фиброза миокарда, медиана объема фиброза миокарда составила 14,5%.
За период наблюдения (медиана наблюдения 36 месяцев) имплантируемые устройства (ИКД/СРТ-Д) были установлены 16 (21,1%) пациентам, оправданные шоки отмечались у двух из них; трое пациентов умерли, из них ВСС наступила у одного пациента с CRT-D-терапией, которая оказалась неэффективной в купировании устойчивой ЖТ. Троим пациентам успешно проведена ортотопическая трансплантация сердца (ОТС).
В результате проведения секвенирования методом NGS у 26 из 30 пробандов с аритмогенным фенотипом НКМ 86,7%) об-
Таблица 1. Клиническая характеристика пациентов с клинически значимыми желудочковыми аритмиями Table 1. Clinical characteristics of patients with clinically significant ventricular arrhythmias
|
Характеристика |
Всего, n=76 |
|
Возраст вступления в исследование, лет |
42 [18; 69] |
|
Мужской пол, n (%) |
48/28 (63,2/36,8) |
|
ЧСС, уд/мин |
69 [58,5; 80] |
|
Продолжительность QRS, мс |
110 [99, 133] |
|
Длительность корригированного интервала QT, мс |
437 [409; 480] |
|
ЖЭ более 500 в сутки, n (%) |
50 (65,8) |
|
Наличие ФП, форма постоянная, n (%) |
34 (44,7) |
|
Наличие неустойчивой ЖТ в сутки, n (%) |
54 (71,1) |
|
Наличие устойчивой ЖТ в сутки, n (%) |
11 (14,5) |
|
ФК СН NYHA 0, n (%) I, n (%) II, n (%) III, n (%) |
2 (2,6) 17 (22,4) 37 (48,7) 20 (26,3) |
|
ФВ ЛЖ, % |
41 (17; 80) |
|
Число пациентов ФВ ЛЖ <40%, n (%) |
35 (46) |
|
Характеристика |
Всего, n=76 |
|
Число пациентов с ФВ ЛЖ >40%, n (%) |
41 (54) |
|
ЛП, мм |
41 [28; 70] |
|
КДР ЛЖ, мм |
61 [38; 89] |
|
КСР ЛЖ, мм |
49 [26; 75] |
|
КДО ЛЖ, мл |
177 [40; 470] |
|
КСО ЛЖ, мл |
98 [9; 384] |
|
Число пациентов с фиброзом миокарда, n (%) |
37 (48,7) |
|
% объема фиброза миокарда |
14,35 [0,63; 50] |
|
Фенотип НКМ+ДКМП, n (%) |
41 (53,9) |
|
Фенотип НКМ+ГКМП, n (%) |
8 (10,5) |
|
Изолированный НКМ, n (%) |
24 (31,6) |
|
Фенотип НКМ+ВПС, n (%) |
3 (3,9) |
|
Имплантируемые устройства: |
16 (21,1) |
|
ИКД, n (%) |
7 (9,3) |
|
CRT-D/ИКД, n (%) |
9 (11,8) |
|
ОТС, n (%) |
3 (3,9) |
|
Смерть, n (%) |
3 (3,9) |
Примечания: данные представлены в виде n (%), M±SD, Me [LQ; UQ]; ЖЭ – желудочковая экстрасистолия; ФП – фибрилляция предсердий; ЖТ – желудочковая тахикардия; ФК СН – функциональный класс сердечной недостаточности; ФВ ЛЖ- фракция выброса левого желудочка; ЛП – левое предсердие; КДР ЛЖ – конечно-диастолический размер; КСР ЛЖ – конечно-систолический размер; КДО ЛЖ – конечно-диастолический объём левого желудочка; КСО ЛЖ – конечносистолический объём левого желудочка; НКМ – некомпактный миокард; ДКМП – дилатационная кардиомиопатия; ГКМП – гипертрофическая кадиомиопатия; ВПС – врождённый порок сердца; ОТС – ортотопическая трансплантация сердца. Notes: data are presented as n (%), M±SD, Me [LQ; UQ]; VPB – ventricular premature beats; AF – atrial fibrillation; VT – ventricular tachycardia; FC HF – functional class of heart failure; LV EF - left ventricular ejection fraction; LA – left atrium; LV EDD – left ventricular end-diastolic diameter; LV ESD – left ventricular end-systolic diameter; LV EDV – left ventricular end-diastolic volume; LV ESV – left ventricular end-systolic volume; NCCM – non – compaction cardiomapathy; DCM – dilated cardiomyopathy; HCM – hypertrophic cadiomyopathy; CHD – congenital heart disease; OHT – orthotopic heart transplantation.
наружено 40 изменений нуклеотидной последовательности в 27 генах. На рисунке 1 представлено распределение выявленных мутаций в генах, ответственных за синтез саркомерных, структурных белков и субъединиц ионных каналов. Доля мутаций в генах саркомерных протеинов составила 26,9%, а в генах белков ионных каналов оказалась равной 23,1%. На долю нуклеотидных изменений в генах, кодирующих структурные белки, пришлось 11,5%. Две и более мутаций в различных генах обнаружены в 38,5% случаях, причем в 30,8% аминокислотные изменения затрагивали белки разных функциональных классов.
Доля миссенс-мутаций была наибольшей и равнялась 70%, остальные 12 нуклеотидных вариантов были представлены де-лециями без сдвига рамки считывания (5), изменениями в интроне, нарушающими сплайсинг (2), антисмысловыми мутациями (3) и дупликациями со сдвигом рамки считывания (2).
В таблице 2 представлены выявленные в результате секвенирования мутации в генах, кодирующих саркомерные белки: 3 мутации в гене MYH7 и по одной мутации в генах MYBPC3, TTN и NEXN .
Патогенные мутации составили 28,6% (2 из 7); варианты с неопределенной клинической значимостью (variant of uncertain significance, VUS) – 57,1% (4 из 7) и 1 из мутаций в гене MYH7 была новой (табл. 2). Пациенты с заменами VUS в генах MYH7, MYBPC3, NEXN и LDB3 имели желудочковые аритмии, потребовавшие имплантации ИКД/CRT-D. 2 пациента впоследствии умерло, один (код 4) скончался вследствие развития тромбоэмболических осложнений, а второй пробанд (код 6) с наличием желудочковой аритмии и полной блокады левой ножки пучка Гиса – из-за отсутствия эффективности в купировании устой- чивой ЖТ выполненной CRT-D-терапии. Пациенты (код 1, 2) с мутациями в гене MYH7 и пациент (код 5) с нонсенс-мутацией в гене TTN на данный момент характеризовались отсутствием жизнеугрожающих аритмических событий. В исследованиях последних лет мутации, ведущие к возникновению усеченных вариантов титина, считаются самой частой причиной возникновения НКМ [17]. При этом в нескольких независимых исследованиях на основе семейного скрининга показано, что усеченные варианты TTN часто ассоциируются с аритмогенными событиями и имеют риск неблагоприятного исхода [18,19].
В таблице 3 представлены данные генотипирования пациентов, имеющих аминокислотные изменения в белках ионных каналов.
У 28,8% (8 из 28) пациентов с НКМ были выявлены 10 генетических вариантов в 8 генах, кодирующих белки ионных каналов и ассоциированных с ними протеинов: KCNQ1 (1), KCNH2 (2), KCNE1 (1), SCN2B (1), RYR2 (1), CACNA1C (1), HCN4 (1) и DPP6 (2). Патогенные мутации составили 10,0% (1 из 10), остальные 90,0% классифицировались как VUS. У 2 из 8 пациентов (код 10 и 12) было выявлено сочетание двух редких вариантов в генах ионных каналов. У всех пациентов (код 9 и 10) с мутациями в гене KCNH2 аритмические события потребовали имплантацию ИКД/CRT-D. Интересно отметить, что удлинения интервала QT на сериях ЭКГ ни у одного из пациентов с мутациями в генах, кодирующих белки ионных каналов, зарегистрировано не было.
В исследовании Miszalski-Jamka et al. [20] в когорте из 190 пациентов с НКМ при секвенировании всего экзома (WES) обнаружено значительное количество генетических вариантов, контролирующих ионные каналы: KCNH2, SCN5A, RYR2, из которых
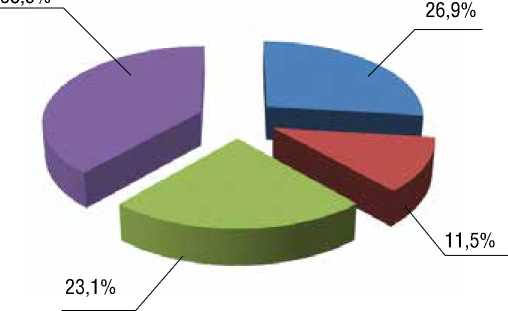
Рисунок 1. Распределение мутаций в генах у пациентов с аритмическим фенотипом НКМП
Figure 1. Mutations distribution in patients’ genes with arrhythmic phenotype of NCCM

Гены саркомерных белков
Гены структурных белков
Гены белков ионных каналов
Две и более мутации
Таблица 2. Выявленные мутации в генах саркомерных белков у пациентов с НКМ
Table 2. Identified mutations in sarcomeric protein genes in patients with NCCM
|
Код пациента |
Ген |
Нуклеотидная замена, Rs |
Аминокислотная замена |
Статус мутации |
События и исходы |
|
1 |
MYH7 |
с.4010G>A, rs368575559 |
p.Arg1337Gln |
VUS |
_ |
|
2 |
MYH7 |
c.2785_2787GAG rs397516172 |
p.Glu931del |
Рathogenic |
_ |
|
3 |
MYH7 |
c. 1169T>G |
p.Leu390Arg |
New |
ИКД |
|
4 |
MYBPC3 |
c.3763G>A, rs727503167 |
p.Ala1255Thr |
VUS |
CRT-D, летальный исход |
|
5 |
TTN |
c.53488G>T, rs759231562 |
p.Gly17830* |
Likely pathogenic |
_ |
|
6 |
NEXN |
c.298+1G>C, rs780476936 |
нарушение сплайсинга |
VUS |
CRT-D, летальный исход |
|
7 |
LDB3 |
c.665G>A, rs139922045 |
p.Ala222Thr |
VUS |
ИКД |
KCNH2 и SCN5A ассоциированы с синдромом удлиненного интервала QT. Несмотря но то, что механизм, с помощью которого мутации в генах ионных каналов могут вызывать НКМ, неясен, данные исследования показывают преобладание редких вариантов в этих генах по сравнению с имеющимися популяционными базами данных. В исследовании Richard et al. [21] в когорте из 95 неродственных пациентов с НКМ сообщалось о высокой доле пациентов с патогенными мутациями в гене HCN4. Среди этих пациентов у 3/5 была выявлена брадикардия (одному пациенту был имплантирован ЭКС). В нашей когорте жизнеугрожающих аритмических событий у пациента (код 12) с вариантами в генах HCN4 и SCN2B зарегистрировано не было. У 2 пробандов (код 13, 14) была выявлена одинаковая делеция в гене DPP6 . В литературе описана мутация в этом гене у пациента с фибрилляцией желудочков (ФЖ), многократно рецидивирующей вопреки многочисленным фармакологическим и абляционным вмешательствам [22]. У 2 наблюдаемых нами пациентов с одинаковой делецией в гене DPP6 в настоящий момент регистрируются злокачественные желудочковые аритмии (индекс эктопии – 21,7%) и проводится медикаментозное лечение соталолом 160 мг/сутки.
Среди 3 пациентов (10,7%) с мутациями в генах, кодирующих структурные белки, имплантация CRT-D и ОТС проведена двум пробандам с нуклеотидными вариантами в генах PKP2 и ACTN2 (см. табл. 4).
В литературе мутации в гене PKP2 представлены, как наиболее частая генетическая причина развития АДПЖ [22]. Имеются также единичные сообщения о связи мутаций в гене PKP2 с развитием НМЛЖ. Так, в одном из исследований описана гомозиготная делеция (hg19, chr12: 32,936,266_33,056,189) размером 120 kb, захватывающей весь ген PKP2, у брата и сестры с диагнозом некомпактного миокарда левого и правого желудочков [23]. Отсутствие гена оказалось летальным: один ребенок умер пренатально, второй – в возрасте 12 дней. Однако у гетерозиготных по данной делеции родителей, состоявших в близкородственном браке, ультразвуковое исследование сердца было без отклонений. У наблюдаемого нами 53-летнего пациента с мутацией c.1892A>C, p.Tyr631Cys в гене РКР2 обнаружена НКМ, подтвержденная МРТ-критериями с дилатацией полости ЛЖ (КДР 74 мм, КСР 65 мм), снижением ФВ ЛЖ до 23% и наличием частой желудочковой экстрасистолии (12800 за сутки) с пробежками неустойчивой ЖТ и полной блокадой левой ножки пучка Гиса. Пациенту было имплантировано CRT-D и назначена стандартная терапия ХСН. В динамике наблюдения клинические проявления СН значительно уменьшились, регистрировались уменьшение размеров ЛЖ (КДД 62 мм, КСД 55 мм) и повышение ФВЛЖ до 30%.
В исследуемой когорте отмечена большая доля пациентов (30,8%) с несколькими нуклеотидными вариантами в двух или более генах (табл. 5), кодирующих белки с различной функциональной направленностью. У 8 пациентов выявлено 20 нуклеотидных вариантов: 2 патогенных мутации, 15 вариантов с неопределенной значимостью и 3 новых замены.
У 5 из 8 пациентов (62,5%) с несколькими мутациями в различных генах наблюдались неблагоприятные события и исходы. Жизнеугрожающие желудочковые аритмические события с необходимостью имплантации ИКД были зарегистрированы у 3 пробандов с различным сочетанием мутаций в саркомерных, структурных генах и генах ионных каналов. Так, у 65-летнего пациента (код 23) с устойчивой ЖТ и синкопальными состояниями, которому был имплантирован ИКД, обнаружены четыре генетический варианта с неустановленной клинической значимостью. У 51-летней пациентки (код 22) с частыми пароксизмами неустойчивой ЖТ и оправданными срабатываниями ИКД при генотипировании обнаружены 3 генетических варианта VUS
Таблица 3. Выявленные мутации в генах ионных каналов и ассоциированных с ними белков у пациентов с НКМ Table 3. Identified mutations in the ion channels genes and associated proteins in patients with NCCM
|
Код пациента |
Ген |
Нуклеотидная замена, Rs |
Аминокислот-ная замена |
Статус мутации |
События и исходы |
|
8 |
KCNQ1 |
c.477+1G>A, rs762814879 |
нарушение сплайсинга |
Pathogenic |
- |
|
9 |
KCNH2 |
c.3107G>A, rs199473022 |
p.Gly1036Asp |
VUS |
ИКД |
|
10 |
KCNH2 |
c.1633C>T, rs143512106 |
p.Arg545Cys |
VUS |
CRT-D |
|
KCNE1 |
c.253G>A, rs1805128 |
p.Asp85Asn |
Risk factor |
||
|
11 |
CACNA1С |
c.2807Т>G |
p.Phe936Cys |
VUS |
_ |
|
12 |
HCN4 |
c.3461G>A, rs145862018 |
p.Arg1154Gln |
VUS |
— |
|
SCN2B |
c.625-626delinsCC, rs1064796044 |
p.Asn209Pro |
VUS |
||
|
13 |
DPP6 |
c.166_167insGCG, rs926747893 |
p.Arg56delins ArgGly |
VUS |
_ |
|
14 |
DPP6 |
c.166_167insGCG, rs926747893 |
p.Arg56delins ArgGly |
VUS |
_ |
|
15 |
RYR2 |
c.12272C>T, rs794728783 |
p.Ala4091Val |
VUS |
_ |
Таблица 4. Выявленные мутации в генах структурных белков у пациентов с НКМ
Table 4. Identified mutations in structural protein genes in patients with NCCM
Наши результаты указывают на то, что в 62,5% случаев наличие мутаций в двух или трех генах привело к жизнеугрожающим желудочковым аритмиям (неустойчивой и устойчивой ЖТ), имплантации КД и неблагоприятным исходам, что позволяет утверждать о кумулятивном эффекте выявленных генетических вариантов, который проявляется злокачественным фенотипом заболевания. С другой стороны, одна из мутаций у этих пациентов идентифицирована в гене саркомерных белков. Этот факт указывает на большую значимость в развитии злокачественных желудочковых аритмий именно мутаций в генах саркомерных белков: половине пациентов даже только с одной мутацией в этих генах установлены имплантируемые устройства (ИКД/ СРТ-Д) (табл. 1), тогда как пациентам с мутациями в других генах имплантация КД требовалась реже (табл. 3-5). Полученные данные являются предварительными и требуют дальнейшего углубленного изучения.
Таблица 5. Множественные мутации у пациентов с НКМ в генах, кодирующих белки с различной функциональной направленностью Table 5. Multiple mutations in patients with NCCM in genes encoding proteins with different functions
|
Код пациента |
Ген |
Нуклеотидная замена, Rs |
Аминокислотная замена |
Статус мутации |
События и исходы |
|
19 |
MYBPC3 |
c.1037G>A, rs397515883 |
p. Arg346His |
VUS |
— |
|
ELN |
c.2032G>A, rs375579231 |
p.Gly678Ser |
VUS |
||
|
MYBPC3 |
c.1037G>A, rs397515883 |
p. Arg346His |
VUS |
_ |
|
|
20 |
DPP6 |
c.166_167insGCG, rs926747893 |
p.Arg56delins ArgGly |
VUS |
|
|
21 |
MYBPC3 |
c.1037G>A, rs397515883 |
p. Arg346His |
VUS |
Летальный исход |
|
TGFB2 |
c.52G>T, rs886045975 |
p.Ala18Ser |
New |
||
|
MYBPC3 |
с.3697C>T, rs397516037 |
p.Gln1233* |
Рathogenic |
ИКД |
|
|
22 |
DTNA |
c.1663A>G, rs779045040 |
p.Asn555Asp |
VUS |
|
|
JPH2 |
c.1225A>G |
p.Asn409Asp |
New |
||
|
NOTCH1 |
c.823G>A, rs371333249 |
p.Gly275Ser |
VUS |
||
|
TTN |
c.12283G>T |
p.Glu4095* |
New |
ИКД |
|
|
23 |
RYR2 |
c.12665_12667delAGA, rs794728838 |
p.4222_4223del |
VUS |
|
|
DSP |
c.2887C>T, rs779931043 |
p.Leu963Phe |
VUS |
||
|
TMPO |
c.629T>A, rs781269460 |
p.Val210Glu |
VUS |
||
|
24 |
TCAP |
c.208C>T, rs775636212 |
p.Arg70Trp |
VUS |
ИКД |
|
KCNQ1 |
c.1450G>A, rs147445322 |
p.Asp484Asn |
VUS |
||
|
JPH2 |
c. 1720A>G, rs773306912 |
p.Thr574Ala |
VUS |
_ |
|
|
25 |
AKAP9 |
c.11714T>C, rs77447750 |
p.Met3905Thr |
Benign (1); Likely benign (1); VUS (3) |
|
|
26 |
PLN |
c.26_29dupCTCG |
p.Thr11fsLeuX10 |
Рathogenic |
ОТС |
ЗАКЛЮЧЕНИЕ
В статье представлено изучение группы пациентов с НКМ с преобладающим аритмическим фенотипом, которую следует рассматривать как группу повышенного риска жизнеугрожающих желудочковых тахиаритмий и ВСС. Результаты генотипирования показали превышение среди этой когорты лиц пациентов с нуклеотидными вариантами (86,7%) по сравнению с общей группой пациентов с НКМ (59%) [20]. У 26 из 30 пациентов выявлено 40 изменений нуклеотидной последовательности в 27 генах, ассоциированных с развитием различных кардиомиопатий. Большинство мутаций зафиксировано в генах, кодирующих саркомерные белки и субъединицы ионных каналов. В 38,5% случаев выявлена не одна, а две или более редких мутаций, причем в 30,8% аминокислотные изменения затрагивали белки разных функциональных классов. Наибольшая частота неблагоприятных событий и исходов наблюдалась у пациентов с несколькими мутациями в генах, кодирующих белки разных функциональных классов (62,5%), при этом одна из мутаций локализовалась в генах саркомерных белков: MYBPC3, TTN, PLN и TCAP . В целом в группе пациентов с имплантируемыми устройствами (ИКД/СРТ-Д) мутации в генах саркомерных белков наблюдались чаще, чем в других генах.
Генетические особенности пациентов наряду с клиническими характеристиками являются маркерами высокого риска развития жизнеугрожающих желудочковых аритмий и могут дополнительно использоваться для прогнозирования неблагоприятных событий у пациентов с НКМ, а также для ранней диагностики заболевания у их ближайших родственников. Определение генетической составляющей открывает новые возможности для персонализации стратификации рисков пациентов с НКМ.
Список литературы Генетические причины аритмического фенотипа некомпактной кардиомиопатии
- Elliott P, Andersson B, Arbustini F, et al. Classification of the cardiomyopathies: a position statement from the European society of cardiology working group on myocardial and pericardial disease. Eur Heart J 2008;29 (2); 270-6. D0l:10.1093/eurheart/ehm342.
- Kovacevic-Preradovic T, Jenni R, Oechslin E et al. Isolated left ventricular noncompaction as a cause for heart failure and heart transplantation: a single center experience. Cardiology. 2009; 112: 158-64. DOI: 10.1159/000147899.
- van Waning J, Caliskan K, Michels M, et al. Cardiac phenotypes, genetics, and risk familian noncompaction cardiomyopathy. J Am Coll Cardiol. 2019; 73:1601-11. DOI: 10.1016/j.jacc.2018.12.085.
- Jefferies J. Barth Syndrome. Am J Med Genet C Semin Med Genet. 2013; 163 С: 198 -205. DOI: 10.1002/ajmg.c.31372.
- Timolo A.Z, Nguyen T, et al. Spectrum of Cardiac Arrhythmias in isolated ventricular n0n-compaction. The J of innovations in cardiac rhythm management.2018:2777-2783. DOI: 10.19102/ icrm. 2017.080701.
- Muser D, Liang J, Witsehey W, et al. Ventricular arrhythmias associated with left ventricular noncompaction: electrophysiological characteristics, mapping, and ablation. Heart Rhythm. 2017; 14 (2): 166 - 75. DOI: 10.1016/j.hrhythm 2016.11.01.14.
- Kayvanpour E, Sedaghat-Hamedani F, Gi W, et al. Clinical and genetic insights into non-compaction: a meta-analysis and systematic review on 7598 individuals. Clin Res Cardiol. 2019. DOI: 10.1007/s00392-019-01465-3.
- Haugaa KH, Dan GA, Iliodromitis K. Management of patients with ventricular arrhythmias and prevention of sudden cardiac death-translating guidelines into practice: results of the European Heart Rhythm Association survey. Europace. 2018;20:f249-f253. DOI: 10.1093/ europace/euy112.
- Towbin JA, McKenna WJ, Abrams DJ. 2019 HRS expert consensus statement on evaluation, risk stratification, and management of arrhythmogenic cardiomyopathy. Heart Rhythm. 2019;16(11):e301-e372. DOI: 10.1016/j.hrthm.2019.05.007.
- Towbin J, Lorts A, Jefferies J. Left ventricular non-compaction cardiomyopathy. The Lancet, 2015; 386: 813-25. DOI: 10.1016/S0140-6736(14)61282-4.
- Towbin J, McKenna W, Abrams D, et al. 2019 HRS expert consensus statement on evaluation, risk stratification, and management of arrhythmogenic cardiomyopathy: Executive summary. Heart Rhythm. 2019;16(11):e373-e407. DOI: 10.1016/j.hrthm.2019.09.019.
- van Waning J, Moesker J, Heijsman D, et al. Systematic Review of Genotype-Phenotype Correlations in Noncompaction Cardiomyopathy. J Am Heart Assoc. 2019;8(23):e012993. DOI: 10.1161/JAHA.119.012993.
- Jenni R, Oechslin E, Schneider J, et al. Echocardiography and pathoanatomical characteristics of isolated left ventricular non-compaction: a step towards classification as a distinct cardiomyopathy. Heart (British Cardiac Society). 2001;86 (6):666-71. DOI: 10.1136/ heart.86.6.666.
- Petersen SE, Selvanayagam JB, Wiesmann F, et al. Left ventricular non-compaction: in- sights from cardiovascular magnetic resonance imaging. Journal of the American College of Cardiology. 2005;46 (1):101-5. DOI: 10.1016/j.jacc.2005.03.045.
- Jacquier A, Thuny F, Jop B, et al. Measurement of trabeculated left ventricular mass using cardiac magnetic resonance imaging in the diagnosis of left ventricular non-compaction. European Heart Journal. 2010;31 (9):1098-104. DOI: 10.1093/eurheartj/ehp595.
- Wang K, et al. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Research - 2010. - Vol. 38. - P. 164. DOI: 10.1093/nar/gkq603.
- Roberts A, Ware J, Herman D, et al, Integrated allelic transcriptional, and phenomic dissection of the cardiac effects of titin truncations in health and disease. Sci Transl Med. 2015; 7:270 - 6. DOI:10,1126 /Sci transmed.3010134.
- Ware J, Cook S. Role of titin in Cardiomyopathy from DNA Variants to patient's stratification. Nat Rev Cardiol 2017; 15 (4): 241 - 52. DOI: 101038/ nrcardio.2017.190.
- Sedaghat-Hamedani F, Haas J, Zhu F, et al. Clinical genetics and outcome of left ventricular non-compaction cardiomyopathy. Eur Heart J. 2017;38(46):3449-3460. DOI: 10.1093/eurheartj/ehx545
- Miszalski-Jamka K, Jefferies JL, Mazur W, et al. Novel Genetic Triggers and Genotype-Phenotype Correlations in Patients with Left Ventricular Noncompaction. Circulation: Cardiovascular Genetics. 2017;10(4):e001763. DOI:10.1161/CIRCGENETICS.117.001763.
- Richard P, Ader F, Roux M, et al. Targeted panel sequencing in adult patients with left ventricular non-compaction reveals a large genetic heterogeneity. Clin Genet. 2019;95:356-367. DOI: 10.1111/cge.13484.
- Trenkwalder T, Deisenhofer J, Hadamitzky M, et al. Novel frome-scift mutations in PKP2 associated with arrhythmogenic right ventricular cardiomyopathy. A case report. BMC Med Genet.2015; 16:117. DOI: 10.1186/s12881-015-0263-1.
- Ramond F, Janin A, Filipo SD, et al. Homozygous PKP2 associated with neonatal left ventricle noncompaction. Clin Genet. 2017;91(1):126-130. DOI: 10.1111/cge.12780.
- Posch M, Perrot A, Geier C, et al. Genetic detection of arginine 14 in causes dilated cardiomyopathy with attenuated electrocardiographic R amplitudes. Heart Rhythm. 2009; 6: 480-6. DOI: 10.1016/j. hrthm.2009.01.016.
- Van Rijsingen J, van der Zwaag P, Groeneweg J, et al. Outome in phospholamban R14del Carries Result of a large multicenter cohort study. Circ Cardiovasc Genet. 2014; 8: 1942-48. DOI 10.1161/ CIRGENETICS.113.000374.
- Haghighi K, Kolokathis F, Pater L, et al. Human phospholamban null results in lethal dilated cardiomyopathy revealing a critical difference between mouse and human. J Clin Invest. 2003;111(6):869-876. DOI:10.1172/ JCI17892.
