Генетический спектр семейной и спорадической дилатационной кардиомиопатии: аритмические фенотипы, ассоциированные с мутациями в гене ламина A/C (LMNA)
")
Автор: Вайханская Т. Г., Сивицкая Л. Н., Курушко Т. В., Левданский О. Д., Даниленко Н. Г.
Журнал: Евразийский кардиологический журнал @eurasian-cardiology-journal
Рубрика: Оригинальные статьи
Статья в выпуске: 2, 2023 года.
Бесплатный доступ
Цель исследования. Изучение диагностической значимости каскадного семейного скрининга и спектра генетических вариантов у пациентов с семейной и спорадической дилатационной кардиомиопатией (ДКМП), оценка клинических исходов и сравнительный анализ 5-летней бессобытийной выживаемости.
Дилатационная кардиомиопатия, каскадный скрининг, генетические варианты, ген ламина a/c (lmna), кумулятивная выживаемость, аритмический риск
Короткий адрес: https://sciup.org/143180186
IDR: 143180186 | УДК: 616.12-007.61 | DOI: 10.38109/2225-1685-2023-2-26-37
Genetic spectrum of familial and sporadic dilated cardiomyopathy: arrhythmic phenotypes associated with mutations in the lamin A/C (LMNA) gene
Purpose. To study the diagnostic value of cascade family screening and the spectrum of genetic variants in patients with familial and sporadic DCM, assess clinical outcomes and comparative analysis of 5-year event-free survival.Materials and methods. The study included 156 unrelated patients with verified DCM. All patients (aged 46 [34; 57] years; 125 (80%) male; LVEF 31 [24; 38]%; LV EDD 68 [61; 74] mm; follow-up period - 77 [59; 108] months) a complex of clinical and instrumental studies (ECG, ECHO, HM, MRI), cascade family screening with genetic testing (NGS+Sanger) and segregation analysis were performed.Results. Criteria for familial DCM were identified in 73 (46.8%) probands. The genetic cause of DCM was identified in 47 (64,4%) familial cases, while for sporadic form DCM pathogenic variants were detected in 19 (22,9%) patients. The dominant mutations were truncating variants in the titin gene (10,9%) and variants in the lamin A/C (LMNA) gene - 8,33%. As a result of the evaluation of cumulative event-free survival (Kaplan-Meier curves), LMNA carriers showed the poor 5-year prognosis for ventricular tachyarrhythmic events (x2=39.9; p=0,0001) and composite adverse outcomes (x2=12.1; p=0.001). Probands who had a familial DCM (log rang x2=38.5; p=0,0001) showed the worst prognosis and low cumulative survival when compared with patients of the sporadic DCM.Conclusion. Cascade clinical family screening and genetic testing in the DCM cohort increased the level of diagnosis of familial DCM from 4.5% to 46.8%. Associations of LMNA mutations with life-threatening tachyarrhythmias are defined as prognostically significant, that confirms the important role of genetic stratification of arrhythmic risk.
Текст научной статьи Генетический спектр семейной и спорадической дилатационной кардиомиопатии: аритмические фенотипы, ассоциированные с мутациями в гене ламина A/C (LMNA)
Данная статья распространяется на условиях «открытого доступа», в соответствии с лицензией CC BY-NC-SA 4.0 («Attribution-NonCommercial-ShareAlike» / «Атрибуция-Некоммерчески-Сохранение Условий» 4.0), которая разрешает неограниченное некоммерческое использование, распространение и воспроизведение на любом носителе при условии указания автора и источника. Чтобы ознакомиться с полными условиями данной лицензии на русском языке, посетите сайт:
This is an open access article distributed under the terms of the Creative Commons Attribution-NonCommercial-ShareAlike 4.0 International (CC BY-NC-SA 4.0) License , which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Дилатационная кардиомиопатия (ДКМП) относится к спектру гетерогенных заболеваний миокарда, характеризующихся дилатацией камер сердца и их систолической дисфункцией (при отсутствии других известных причин – артериальной гипертензии, клапанных пороков сердца, врожденных или ишемических заболеваний сердца). Этиология ДКМП также отличается разнообразием и включает генетические мутации, инфекции, воспалительные процессы, аутоиммунные заболевания, воздействие токсинов и эндокринные или нейромышечные расстройства [1].
Генетическое тестирование в настоящее время позволяет идентифицировать патогенные варианты примерно у 15–25% пациентов с идиопатической ДКМП и у 20–60% пациентов с семейной ДКМП [2, 3]. Однако семейная природа ДКМП не всегда является очевидной. В клинической практике ДКМП чаще диагностируют как спорадическое заболевание, так как члены семьи редко располагают точной информацией о наследственных заболеваниях у родственников. В случае, если семейный анамнез тщательно не изучен клиницистом, наследственное заболевание ошибочно классифицируют как спорадическую форму ДКМП. Однако некоторая часть спорадических случаев ДКМП также имеет генетическую этиологию, обусловленную мутациями de-novo (впервые возникшие варианты, в отличие от унаследованных). При семейной и спорадической ДКМП исследователи часто обнаруживают мутации сходного спектра генов. Общность генетической этиологии указывает на то, что некоторые спорадические случаи в действительности могут быть нераспознанной формой семейной ДКМП [3].
Среди генетических причин ДКМП хорошо изучены мутации в более 150 генах, приводящих к синтезу дефектных белков в различных клеточных структурах (саркомер, ядерная оболочка, Z-диск, мембрана, ионные каналы, сигнальные пути) [2, 3]. Однако лишь для 19 генов международными экспертами получены строгие доказательства ведущей молекулярно-генетической роли в развитии заболевания (высокий уровень определен для генов BAG3, DES, DSP, FLNC, LMNA, MYH7, PLN, RBM20, SCN5A, TNNC1, TNNT2, TTN и умеренный уровень доказательств – для генов ACTC1, ACTN2, JPH2, NEXN, TNNI3, TPM1, VCL ), а остальные гены имеют пока ограниченные доказательства моногенной связи с ДКМП [4]. По данным многоцентровых исследований мутации в гене самого большого белка титинa ( TTN ), приводящие к укорочению белка, – TTNtv (от англ. truncating variants of TTN ), являются наиболее частой причиной ДКМП. В 19–25% случаев они ассоциированы с семейной формой заболевания и в 11–18% связаны с развитием спорадических форм [5]. Другой наиболее распространенный ген, кодирующий белки ядерной пластинки ламины – ламин A/C ( LMNA ), встречается в 4–11% случаев ДКМП, в том числе в 35–40% случаев аритмических фенотипов с нарушениями проводимости [6]. Ламин А и ламин С представляют собой две изоформы протеина, кодируемые одним геном LMNA , расположенным на хромосоме 1q21.2-q21.3. Тип наследования кардиальных ламинопатий – аутосомно-доминантный, с практически полной пенетрантностью к 7-му десятилетию [6, 7].
К настоящему времени зарегистрировано более 500 патогенных вариантов LMNA, вызывающих широкий спектр заболеваний, начиная от преждевременного старения и заканчивая метаболическими и скелетно-мышечными нарушениями [7]. Кардиоламинопатии – это редкие заболевания с гетерогенными кардиальными проявлениями, которые варьируют от легких морфоструктурных изменений (легкая или умеренная дилатация левого желудочка (ЛЖ) с дефектами проводящей системы или суправентрикулярной аритмией) до тяжелой систолической дисфункции, жизнеопасных желудочковых тахиаритмий (ЖТА) с развитием внезапной сердечной смерти (ВСС). Электрические нарушения часто предшествуют развитию структурных изменений, нередко ВСС развивается как первое проявление кардиоламинопатии при сохраненной систолической функции ЛЖ [6]. Бради- и тахиаритмии, нарушения проводимости обычно предшествуют развитию ДКМП от нескольких лет до десятилетий [7]. При манифестации ДКМП суправентрикулярные тахиаритмии выявляются чаще, чем жизнеопасные ЖТА [8]. LMNA-ассоциированные кардиомиопатии характеризуются неблагоприятным прогнозом, наиболее агрессивным клиническим течением и высокой частотой серьезных кардиальных событий [6–8].
С клинической точки зрения, крайне важно своевременно выявить семейный тип наследования ДКМП, в особенности у потомства в период досимптомной стадии болезни, для принятия комплекса превентивных мер (отстранение от спорта, модификация факторов риска, профилактика инфекций, планирование семьи).
ЦЕЛЬ ИССЛЕДОВАНИЯ
Изучение диагностической значимости каскадного семейного скрининга и анализ спектра генетических вариантов у пациентов с семейной и спорадической формой ДКМП, оценка клинических исходов и сравнение 5-летней кумулятивной бессобытийной выживаемости были целью настоящего исследования.
МАТЕРИАЛЫ И МЕТОДЫ
В исследование включили 156 неродственных пациентов с верифицированной ДКМП, обследованных на базе Республиканского научно-практического центра «Кардиология» (Минск, Беларусь) в период с 2013 по 2021 гг. Первый этап клинического скрининга включал подробный сбор анамнеза и построение родословной (конструкция генеалогического древа) у каждого пробанда с диагнозом ДКМП; второй этап каскадного скрининга – клиническое обследование пробандов и всех близких родственников (родители, братья, сестры, дети) с оценкой физикального статуса, регистрацией электрокардиограммы в 12 отведениях (ЭКГ-12), выполнением эхокардиографии (ЭхоКГ) с тканевой допплерографией и оценкой продольной деформации (GLS) левого желудочка (ЛЖ). Третий этап семейного скрининга включал генетическое тестирование пациентов; при выявлении генетической мутации у пробанда с семейной или спорадической ДКМП проводили генотипирование всех членов семьи 1-й и 2-й линии. Для установления причинно-следственной связи все подтвержденные варианты анализировали специалисты в команде кардиологов, неврологов и генетиков для оценки ге-нотип-фенотипической косегрегации в семье. Таким образом, всем пациентам когорты (возраст 46 [34; 57] лет; средний возраст манифестации ДКМП составил 39 [30; 48] лет; 125 (80,1%) мужчин; фракция выброса (ФВ) ЛЖ 31 [24; 38]%; конечно-диастолический диаметр (КДД) ЛЖ 68 [61; 74] мм) проведен комплекс клинических исследований, включающих: физикальное обследование с детальным изучением семейного анамнеза в 3-х поколениях; нейромышечное исследование с определением уровня сывороточной креатинфосфокиназы (сКФК); ЭхоКГ с оценкой продольной сократимости (GLS); ЭКГ-12 и холтеровское мониторирование (ХМ) 24-48 ч; магнитно-резонансная томография (МРТ) сердца с контрастированием. У пациентов старше 35 лет интактность коронарных артерий была верифицирована с помощью R-контрастной селективной коронароангиографии или компьютерно-томографической ангиографии. Период наблюдения пациентов составил 77 [59; 108] месяцев. Клиническая характеристика пациентов с ДКМП представлена в таблице 1.
Протокол исследования был одобрен локальным Этическим комитетом. При включении в исследование у всех участников (в т.ч. у родственников пробандов) было получено письменное информированное согласие. Исследование выполнялось в соответствии с принципами Хельсинской Декларации.
Геномная ДНК от 156 пациентов была использована для высокопроизводительного секвенирования: ДНК от 126 лиц анализировали (NGS, мультитаргетная кардиопанель 174 генов) на приборе MiSeq System (Illumina Inc., San Diego, CA, US), и у 30 лиц для генетического скрининга образцов ДНК использовали кардио-генетическую панель для таргетного секвенирования 48 генов, ассоциированных c кардиомиопатиями. Патогенность идентифицированных мутаций определяли в соответствии с рекомендациями Американской Коллегии Медицинской Генетики (ACMG) c классификацией вариантов на пять категорий: патогенные (V – pathogenic variant – PV), вероятно патогенные (IV – likely pathogenic variant – LPV), варианты неопределённой клинической значимости (III – variant of unknown significance – VUS), вероятно доброкачественные (II – LBV) и доброкаче- ственные (I – BV) [9]. Для оценки клинической значимости генетических вариантов использовались базы популяционных частот (ESP6500, gnomAD, 1000 Genomes), а также клинико-генетические базы данных CLINVAR, HGMD, LOVD и литературные данные. Патогенность новых генетических вариантов, не зарегистрированных в указанных базах данных, определяли на основании сегрегационного анализа и с помощью соответствующих программ in silico (Revel, Polyphen 2, SIFT, GeneSplicer). Верификацию выявленных мутаций проводили с помощью прямого автоматического секвенирования по методу Sanger.
Генотип-позитивными считали пациентов с патогенными или вероятно патогенными вариантами (PV/LPV) с доказанной клинической значимостью. Генотип-негативную группу составили пациенты с генетическими вариантами, соответствующими критериям доброкачественного значения (LBV/BV) или неизвестной клинической значимости (VUS) без доказательств предполагаемой патогенности. Пробандами считали первых пациентов в семье, направленных на генетическое тестирование при наличии семейного фенотипа ДКМП на основании критериев D. Fatkin [10]: ( а ) если два и более родственника первой линии имели «явные» признаки заболевания или ( б ) при наличии члена семьи, носителя заболевания, с подтвержденнной кар-
Таблица 1. Клиническая характеристика 156 пациентов, включенных в исследование
Table 1. Clinical characteristics of 156 patients included in the study
Cтатистический анализ результатов исследования проводили с помощью биостатистических методов с использованием программы SPSS для Windows (версия 23.0) в соответствии с правилами вариационной статистики для парных и непарных величин, многофакторного анализа, непараметрических методов определения доверительных интервалов, оценки относительного риска, ROC анализа c построением характеристических кривых и анализа кумулятивной бессобытийной выживаемости (с построением кривых Каплан-Мейера).
Количественные параметры представлены в виде медианы (Ме) и квартилей [нижний Q25‰; верхний Q75‰]. Для проверки статистических гипотез о виде распределения был применен критерий Шапиро-Уилкса. Сравнение количественных показателей в группах проводилось с помощью
H-критерия Краскала-Уоллиса. Качественные показатели представлены частотами и процентами в группах. При исследовании таблиц сопряженности применяли критерий хи-квадрат ( χ 2 Пирсона). Статистически значимыми считали различия данных и корреляции между данными при р <0,05.
РЕЗУЛЬТАТЫ
При включении пациентов в исследование диагноз семейной формы ДКМП был установлен у 2-х пробандов (1,28%), ещё 5 (3,21%) пациентов при первичном обследовании вспомнили родственника(ов) с кардиомиопатией и/или раннюю внезапную смерть одного из членов семьи.
При детальном сборе анамнеза и построении генеалогических древ (минимум в 3-х поколениях) у 49 (31,4%) пациентов выявлены потенциальные признаки семейной формы ДКМП. У 23-х (46,9%) из 49 пробандов обнаружены семейные истории необъяснимых преждевременных ВСС. При каскадном клиническом обследовании членов семьи 1-й линии родства еще у 24 (15,4%) пробандов выявлены признаки ДКМП у родствен- ников. Таким образом, семейная форма ДКМП обнаружена у 73 (46,8%) лиц когорты (рис. 1)
В результате генетического исследования у 84 (53,8%) из 156 пациентов с ДКМП выявлено 145 вариантов, из них 70 (48,3%) вариантов согласно критериям AСGM были классифицированы как патогенные или вероятно патогенные (PV/LPV) для 66 (42,3%) носителей, а 75 вариантов соответствовали критериям VUS (в результате каскадного семейного скрининга для этих вариантов не установлена положительная косегрега-ция с заболеванием).
Таким образом, у 66 (42,3%) пациентов установлены генетические причины болезни – патогенные или вероятно патогенные мутации. Компаунд-гетерозиготы и гомозиготные варианты выявлены у 2 носителей, у 2 лиц – дигенные/двойные мутации. В спектре обнаруженных генетических вариантов доминировали мутации в генах белков саркомера (n=28/40%) у 26 пробандов: варианты TTNtv (в т.ч. 2 двойные: n=2/17), MYH7 (n=4), MYBPC3 (n=3), ACTC1 (n=2), MYH6 (n=1). В гене ламина A/C ( LMNA ), кодирующем белки ядерной мембраны, выявлено 12 патогенных вариантов у 13 (8,33%) лиц общей выборки. В генах структурных белков (цитоскелет/Z-диск, ядро) и дес-мосом выявлены патогенные варианты у 13 носителей: BAG3 (n=3), MYPN (n=2), NEXN (n=1), DMD (n=2), RBM20 (n=2), EMD (n=1), DSP (n=2/4; 2 носителя имели по 2 варианта – компаунд гетерозиготы и гомозиготы); в генах транспортных белков ионных каналов, обнаружено 6 PV/LPV у 6-и носителей: SCN5A (n=2), RYR2 (n=1), SCN1B (n=1), TRPM4 (n=1), ABCC9 (n=1). Дилатационный фенотип, ассоциированный с мутациями в генах FLNA (n=1), GATA4 (n=1), XRCC4 (n=1), SLC25A4 (n=1), HADHA (n=1) и LAMP2 (n=3), выявлен у 8-и пациентов с синдромаль-ной патологией и мультисистемными проявлениями.
В группе с семейной формой заболевания генетическая причина ДКМП определена у 47 (64,4%) пробандов из 73, в то время как в группе спорадической ДКМП патогенные мутации выявлены у 19 (22,9%) из 83-х пациентов. Сравнительная структура идентифицированных генетических вариантов у пациентов с семейной и спорадической формой ДКМП в результате классификации мутаций схематически представлена на рисунке 2.
У пациентов с семейной и спорадической ДКМП обнаружено сходство генетического спектра. Однако в представленной когорте LMNA варианты были наиболее частой причиной се-

Анамнез со слов пациента
Рисунок 1. Схема диагностики семейной формы ДКМП
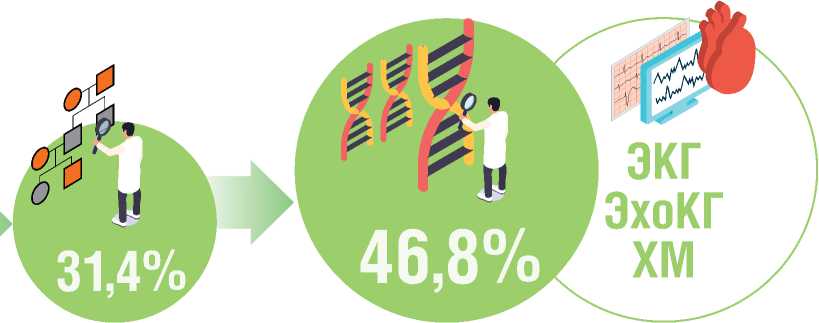
Тщательный сбор анамнеза с построением родословной в 3-х поколениях
Каскадный семейный скрининг с полным клиническим обследованием родственников и генетическим
тестированием
-
Figure 1. Diagnosis scheme for familial DCM
30 ЕВРАЗИЙСКИЙ КАРДИОЛОГИЧЕСКИЙ ЖУРНАЛ, 2, 2023
мейной ДКМП (11/21,6% из всех (n=51) семейных LPV/PV), а TTNtv доминировали у лиц со спорадической ДКМП (9/47,4% вариантов из 19 de-novo мутаций). Шансы выявления гено-тип-фенотипической семейной косегрегации у пациентов с ламинопатиями составили 5,5; у лиц с титинопатиями – 0,89; отношение шансов (OR) составило 6,19 (OR 6,188; 95% ДИ 1,04–36,8; р<0,01). У каждого второго пациента с признаками атриовентрикулярной (АВ) блокады 2–3 степени выявлены мутации в гене LMNA . Нарушения гисиальной проводимости (блокада левой ножки пучка Гиса) реже наблюдались у пациентов с TTNtv генотипом (4/23,5% из 17 TTNtv случаев) в сравнении с носителями LMNA мутаций (7/53,8% из 13; р=0,024) и иными генотипами (19/47,5%).
Среди LMNA вариантов, идентифицированных у пробандов в представленном исследовании (3 frameshift, 1 сплайсинг, 1 non-frameshift и 7 миссенс мутаций), 11 вариантов подтвердились у 16 родственников с положительной семейной гено-тип-фенотипической косегрегацией (табл. 2)
У 4-х (30,8%) из 13 LMNA носителей устойчивые желудочковые тахикардии (уЖТ) и полная АВ блокада у 4 (30,8%) лиц были первыми клиническими проявлениями кардиоламинопа- тии с манифестацией на фоне сохранной систолической функции ЛЖ. У 3-х (23,1%) пробандов ДКМП дебютировала с дисфункции ЛЖ; вследствие быстро прогрессирующей (в среднем 16 мес.) и резистентной к терапии сердечной недостаточности (СН) им потребовалось выполнение трансплантации сердца (ТС). Имплантация устройств с функцией имплантируемого кардиовертера-дефибриллятора (ИКД) выполнена 10-и (76,9%) LMNA носителям. Согласно рекомендациям ESC (2015) [11] 2-м пациентам с нонсенс мутациями в гене LMNA с превентивной целью имплантировали 2-х камерные ИКД (критерии: наличие минимум двух из 4-х следующих факторов риска – (1) неустойчивая ЖТ; (2) ФВ ЛЖ<45%; (3) мужской пол; и (4) нонсенс механизм мутации). В 4-х случаях пациентам с имплантированными ЭКС по поводу полной АВ блокады, после генетического тестирования и идентификации патогенной LMNA мутации, с целью первичной профилактики ВСС была проведена апгрейд-замена ЭКС на устройство сердечной ресинхронизирующей терапии с функцией кардиоверсии-дефибрилляции (СРТ-Д). Ещё 4-м пробандам с целью вторичной профилактики ВСС (после эпизодов синкопе с устойчивой ЖТ/ФЖ) ИКД были имплантированы до установления генетического диагноза ламинопатии.
|
СТРУКТУРА ГЕНОВ |
КОЛИЧЕСТВ 0 5 1 1 |
О ГЕНЕТИ 1 |
ЧЕСКИХ В 5 2 |
АРИАНТО 0 2 |
В 5 |
0 |
ГЕНЫ СЕМЕЙНЫЕ СПОРАДИЧЕСКИЕ С PV/LPV ВАРИАНТЫ DE-NOVO |
|
|
MYH7 |
4■ |
2 |
МYH7 3 1 |
|||||
|
MYBPC3 |
32 |
MYBPC3 2 1 |
||||||
|
MYPN Гены компонентов NEXN саркомера и Z-диска MYH6 TPM1 ACTC1 TTN |
21 12 10 11 21 |
MYPN 2 0 NEXN 1 0 MYH6 1 0 TPM1 1 0 ACTC1 1 1 TTN 8 9 |
||||||
|
17 16 |
||||||||
|
Гены белков внутренней LMNA мембраны ядра EMD |
13 1 |
LMNА 11 2 EMD 1 0 |
||||||
|
10 |
||||||||
|
BAG3 |
30 |
BAG3 2 1 |
||||||
|
Гены компонентов DMD цитоскелета FLNA DES 0 |
20 10 3 |
DMD 1 1 FLNA 1 0 DES - - |
||||||
|
Гены компонентов JUP 0 |
4 |
JUP - - |
||||||
|
десмосом DSP |
DSP 4 0 |
|||||||
|
4 7 |
||||||||
|
SCN5A |
2 |
6 |
SCN5A 2 0 |
|||||
|
TRPM4 |
16 |
TRPM4 1 0 |
||||||
|
RYR2 Гены белков ионных каналов SCN1B SCN2B, CACNAB2 0 KCNE1, KCNH2 0 RYR1 0 |
12 12 2 2 3 |
RYR2 1 0 SCN1B 1 0 SCN2B, - - KCNE1, KCNH2 - - RYR1 - - |
||||||
|
LAMP2 RBM20 SLC25A4 CRYAB 0 |
30 21 10 2 |
LAMP2 2 1 RBM20 2 0 SLC25A4 1 0 CRYAB - - |
||||||
|
Другие гены GATA4 |
13 |
GATA4 0 1 |
||||||
|
GLA 0 ABCC9 XRCC4 1 HADHA 1 |
3 13 0 0 |
GLA - - ABCC9 1 0 XRCC4 0 1 HADHА 1 0 |
||||||
■ PV+LPV ■ VUS
Рисунок 2. Сравнительный спектр выявленных генетических вариантов, ассоциированных с развитием семейной и спорадической ДКМП
-
Figure 2. Comparative spectrum of identified genetic variants associated with the familial and sporadic DCM
EURASIAN HEART JOURNAL, 2, 2023 31
Обоснованные разряды ИКД зарегистрированы у 7 (70%) из 10 пробандов с имплантированными устройствами. Следует отметить, что расчетная медиана 5-летнего риска жизнеугрожающих ЖТА по шкале К. Wahbi et al. [12] с учетом 4-х факторов – пола, АВ блокады, LMNА мутаций, эпизодов неустойчивой ЖТ и ФВЛЖ – составила 40 [23; 67]%, что многократно превысило установленное авторами шкалы «LMNA-risk-vta.fr» пороговое значение высокого риска ( ≥ 7%).
При сравнении исходных данных обнаружено, что пациенты с LMNA генотипом отличалась меньшими размерами ЛЖ – конечно-диастолическим объемом (КДО: Н-критерий Краскала-Уоллиса 13,5; р=0,004) и КДД (Н=9,50; р=0,023), удлинением интервала PR (Н=16,7; р=0,001), а также более юным возрастом манифестации ДКМП (Н=9,62; р=0,022) и дебютом фибрилляции предсердий (ФП: Н=9,93; р=0,019) по сравнению с другими генотипами и генотип-негативными пациентами. Однако частота летальных исходов в группе ламинопатий, несмотря на высокий (84,6%) уровень интервенционного лечения, достигла 46,2% к 6-и годам наблюдения. Клиническая характеристика пробандов с LMNA -ассоциированной ДКМП представлена в таблице 3.
В результате категориального анализа и оценки таблиц сопряженности выявлены ассоциации LMNA генотипа с эпизодами жизнеопасных ЖТА (критерий Пирсона χ2=39,9; р=0,0001). Для оценки и сравнительного анализа неблагоприятных исходов ДКМП в качестве первичной конечной точки (пКТ) были приняты аритмические события – устойчивая желудочковая тахикардия/фибрилляция желудочков (уЖТ/ФЖ), ВСС, успешная сердечно-легочная реанимация (СЛР) и обоснованные разряды ИКД. Композитные кардиоваскулярные неблагоприятные события, такие как: смерть от всех сердечно-сосудистых причин, трансплантация сердца (ТС), уЖТ/ФЖ и СЛР были приняты в качестве комбинированной конечной точки (кКТ).
В общей когорте в 5-летнем периоде наблюдения неблагоприятные клинические события зарегистрированы у 77 лиц с ДКМП: пКТ достигли 30 пациентов, кКТ – 68 лиц (из них 16 пробандов – с летальным исходом), эволюция от пКТ до кКТ наблюдалась у 21 пациента.
У LMNA -носителей в период наблюдения зафиксирован самый высокий уровень неблагоприятных событий: эпизоды устойчивой ЖТ/ФЖ, СЛР или ИКД-терапии зарегистрированы в 92,3% случаев против 10% среди генотип-негативных лиц ( χ 2=39,9; р=0,0001) и 15,1% – в группе других генотипов ( χ 2=25,9; р<0,001). Летальные исходы от всех кардиоваскулярных причин зарегистрированы в 46,2% случаев ламинопатий, уровень летальности значимо превысил частоту смертельных исходов (3,8%) у носителей других генотипов ( χ 2=13,8; р<0,001) и уровень смертности (8,9%) среди генотип-негативных лиц ( χ 2=10,4; р=0,002). У пациентов с LMNA генотипом также значимо чаще регистрировались эпизоды персистирующей ФП ( χ 2=7,19; р=0,008) и АВ блокады 2–3 степени ( χ 2=10,9; р<0,001), у них был выше уровень имплантации СРТ-Д/ИКД устройств в сравнении с группой ге-нотип-негативных лиц ( χ 2=6,02; р=0,015) и носителями других
Таблица 2. Генотип-фенотипическая характеристика 13 пробандов c носительством LMNA мутаций
Table 2. Genotype-phenotypе characteristics of 13 probands carrying LMNA mutations
|
Пробанд с LMNA вариантом (пол) |
Нуклеотидная замена |
Аминокислотная замена |
Экзон |
Нейромышечные нарушения, уровень сывороточной КФК |
Возраст имплантации устройств и/или трансплантации сердца |
Летальный исход (6 лет наблюдения), возраст |
Генотип-фенотипическая косегрегация (+члены семьи), n |
||
|
ЭКС |
ИКД |
ТС |
|||||||
|
1 (муж) |
c.565C>T |
р.R189W |
3 |
- |
- |
36 |
37 |
- |
+2 |
|
2 (жен) |
c.G163C |
р.E55Q |
1 |
Субклинические, ↑ сКФК |
- |
- |
38 |
- |
+1 |
|
3 (муж) |
c.568_569del |
р.L190fs |
5 |
- |
- |
- |
- |
35 |
+2 |
|
4 (муж) |
c.275delT |
р.L92fs |
1 |
Субклинические, ↑ сКФК |
- |
30 |
- |
- |
+3 |
|
5 (жен) |
c.565C>T |
р.R189W |
3 |
- |
- |
25 |
- |
29 |
+1 |
|
6 (муж) |
c.71dupC |
p.T24fs |
1 |
- |
- |
- |
- |
- |
+2 |
|
7 (жен) |
c.1558T>C |
p.R520P |
9 |
ПКМД1Б, ↑ сКФК |
27 |
29 |
30 |
33 |
+1 |
|
8 (жен) |
с.G569C |
p.R190P |
3 |
ПКМД1Б, ↑ сКФК |
- |
21 |
22 |
24 |
- |
|
9 (муж) |
с.343_344delinsTT |
p.E115L |
1 |
- |
- |
40 |
- |
- |
+1 |
|
10 (жен) |
c.745С>G |
p.R249G |
4 |
Субклинические, ↑ сКФК |
22 |
24 |
- |
26 |
+1 |
|
11 (муж) |
c.831_833del |
p.277_278del |
7 |
- |
- |
38 |
- |
- |
+1 |
|
12 (муж) |
c.1583C>G |
p.T528R |
9 |
МДЭД 2, ↑ сКФК |
38 |
45 |
- |
47 |
+1 |
|
13 (муж) |
c.356+1G>A |
Нарушение сплайсинга |
4 |
- |
40 |
44 |
47 |
- |
- |
Примечание: cКФК – сывороточная креатинфосфокиназа ( ↑ –повышение уровня), ИКД – имплантируемый кардиовертер-дефибриллятор; ЭКС – электрокардиостимулятор; ТС – трансплантация сердца; ПКМД 1Б – поясно-конечностная мышечная дистрофия, тип 1Б; МДЭД 2 – мышечная дистрофия Эмери-Дрейфуса, тип 2
Notes: sCPK – serum creatine phosphokinase ( ↑ –increased level), ICD – implantable cardioverter defibrillator; PM – pacemaker; HTx – heart transplant; LGMD 1B – limb-girdle muscular dystrophy, type 1B; EDMD 2 – Emery-Dreyfus muscular dystrophy, type 2
32 ЕВРАЗИЙСКИЙ КАРДИОЛОГИЧЕСКИЙ ЖУРНАЛ, 2, 2023
Таблица 3. Клинико-инструментальная характеристика 13 пациентов с кардиоламинопатиями
Table 3. Clinical and instrumental characteristics of 13 patients with cardiolaminopathies
|
Клинико-инструментальный показатель |
Значение показателя Ме [Q25‰; Q75‰] |
Количество пациентов с расчетным значением, n (%) |
|
Возраст генетического скрининга, лет |
37 [31; 43] |
13 (100) |
|
Возраст манифестации фенотипа ДКМП, лет |
35 [30; 40] |
13 (100) |
|
Возраст дебюта фибрилляции предсердий, лет |
33 [26; 39] |
6 (46,2) |
|
Возраст дебюта атриовентрикулярной блокады 2–3 степени, лет |
31 [25; 36] |
7 (53,8) |
|
Возраст имплантации ЭКС/ИКД и/или трансплантации сердца, лет |
36 [30; 40] |
11 (84,6) |
|
Пол (мужcкой), n (%) |
8 (61,5) |
13 (100) |
|
Семейная форма ДКМП, n (%) |
11 (84,6) |
13 (100) |
|
Желудочковая экстрасистолия в период 24ч ХМ, n |
3872 [886; 13453] |
13 (100) |
|
Фрагментация QRS, случаи n (%) |
7 (53,8) |
13 (100) |
|
Длительность QRS комплекса, мс |
150 [109; 201] |
13 (100) |
|
Максимальная длительность интервала PR, мс |
230 [140; 308] |
7 (53,8) |
|
Фракция выброса ЛЖ в В-режиме, % |
37 [25; 45] |
13 (100) |
|
Конечно-диастолический объем ЛЖ, мл |
170 [138; 205] |
13 (100) |
|
Конечно-диастолический диаметр ЛЖ, мм |
58,7 [55; 62] |
13 (100) |
|
Трансмитральный кровоток, соотношение E/A |
3,4 [2,2; 6,65] |
7 (53,8) |
|
Трансмитральный кровоток, соотношение E/e` |
11 [7; 15] |
7 (53,8) |
|
Фракция выброса ПЖ, % |
46 [38; 52] |
13 (100) |
|
Фракция выброса ЛЖ по данным МРТ, % |
41 [35; 49] |
13 (100) |
|
Признаки фиброза по данным МРТ, случаи (%) |
12 (92,3) |
13 (100) |
|
Значение 5-летнего риска жизнеопасных ЖТА (шкала LMNA-risk-VTA), % |
40 [23; 67] |
13 (100) |
Таблица 4. Сравнительный анализ категориальных параметров и клинических исходов (результаты сравнения пропорций)
Table 4. Comparative analysis of categorical parameters and clinical outcomes (proportion comparison results)
|
Категориальные переменные (наличие или отсутствие признака/ клинического исхода) |
Генотип-негативные лица (группа 1: n=90); n (%) |
LMNA генотип (группа 2: n=13); n (%) |
Другие генотипы (группа 3: n=53); n (%) |
Критерий Пирсона x 2 с поправкой Йейтса для групп сравнения 1_2_3, уровень значимости р |
|
|
Фиброз миокарда |
нет |
65 (72,2%) |
1 (7,7%) |
31 (58,5%) |
гр.1_2: x 2=17,8; р<0,001 |
|
есть |
25 (27,8%) |
12 (92,3%) |
22 (41,5%) |
гр.2_3: x 2=8,85; р=0,003 |
|
|
АВ блокада 2–3 степени |
нет |
79 (87,8%) |
6 (46,2%) |
43 (81,1%) |
гр.1_2: x 2=10,9; р<0,001 |
|
есть |
11 (12,2%) |
7 (53,8%) |
10 (18,9%) |
гр.2_3: x 2=4,98; р=0,026 |
|
|
Фибрилляция предсердий |
нет |
79 (87,8%) |
7 (53,8%) |
44 (83%) |
гр.1_2: x 2=7,19; р=0,008 |
|
есть |
11 (12,2%) |
6 (46,2%) |
9 (17%) |
гр.2_3: x 2=5,06; р=0,025 |
|
|
Имплантация СРТ-Д, ИКД |
нет |
51 (56,7%) |
3 (23,1%) |
38 (71,7%) |
гр.1_2: x 2=6,02; р=0,015 |
|
есть |
29 (32,2%) |
10 (76,9%) |
15 (28,3%) |
гр.2_3: x 2=8,52; р=0,004 |
|
|
Устойчивая ЖТ/ФЖ, |
нет |
80 (90%) |
1 (7,7%) |
45 (84,9%) |
гр.1_2: x 2=39,9; р=0,0001 |
|
СЛР, или ИКД-терапия |
есть |
10 (10%) |
12 (92,3%) |
8 (15,1%) |
гр.2_3: x 2=25,9; р<0,001 |
|
Трансплантация сердца |
нет |
68 (75,6%) |
8 (61,5%) |
39 (73,6%) |
гр.1_2: x 2=0,54; р=0,46 |
|
есть |
22 (24,4%) |
5 (38,5%) |
14 (26,4%) |
гр.2_3: x 2=0,27; р=0,61 |
|
|
Композитная |
нет |
57 (63,3%) |
1 (7,7%) |
30 (56,6%) |
гр.1_2: x 2=12,1; р=0,001 |
|
конечная точка |
есть |
33 (36,6%) |
12 (92,3%) |
23 (43,4%) |
гр.2_3: x 2=8,16; р=0,005 |
|
Летальные исходы |
нет |
82 (91,1%) |
7 (53,8%) |
51 (96,2%) |
гр.1_2: x 2=10,4; р=0,002 |
|
есть |
8 (8,9%) |
6 (46,2%) |
2 (3,8%) |
гр.2_3: x 2=13,8; р<0,001 |
|
Примечание: АВ – атриовентрикулярный, ИКД – имплантируемый кардиовертер-дефибриллятор; СРТ-Д – устройство сердечной ресинхронизирующей терапии с функцией дефибрилляции; ЖТ/ФЖ – желудочковая тахикардия/фибрилляция желудочков Notes: AV – atrioventricular, ICD – implantable cardioverter-defibrillator; СRT-D – device for cardiac resynchronization therapy with defibrillation function; VT/VF – ventricular tachycardia/ventricular fibrillation генотипов (χ2=6,02; р=0,015). Заместительный фиброз миокарда чаще выявляли при контрастной МРТ у пробандов с ламино-патиями (χ2=8,52; р=0,004), чем у лиц с другими генотипами, а также в сравнении с генотип-негативными пациентами (χ2=17,8; р<0,001). Результаты сравнительного анализа морфоструктурных, функциональных признаков и неблагоприятных исходов ДКМП в зависимости от генотипа представлены в таблице 4.
Относительный риск (RR) развития жизнеугрожающих ЖТА у носителей LMNA мутаций составил 6,12 (95% ДИ 3,17–11,8;
чувствительность 60%, специфичность 98%) по сравнению с носителями других генотипов. Риск развития потенциально фатальных ЖТА у пациентов с LMNA генотипом в 8 раз превысил (RR 8,31; 95% ДИ 4,54–13,2; чувствительность 55%, специфичность 99%) аритмический риск генотип-негативных лиц с ДКМП и семикратно (RR 7,33; 95% ДИ 4,63–11,6) – риск потенциально фатальных ЖТА у LMNA -негативных лиц с ДКМП.
Таким образом, в 5-летний период наблюдения носители LMNA мутаций в 6 раз чаще (RR 6,12; 95% ДИ 3,17–11,8) до-
A 1,0
0,8
0,6
cxs
0,2
0,0
0,00 20,00 40,00 60,00 80,00 100,00
наблюдение (месяцы)
1,0
0,8
0,6
0,4
0,2
0,0
0,00 20,00 40,00 60,00 80,00 100,00
наблюдение (месяцы)
Примечание:
Note:
__(1) TTNtv носители __(3) Другие генотипы
__(2) Ген-негативные лица __(4) LMNA носители
__(1) TTNtv carriers
__(2) Gene-negative pts
__(3) Other genotypes
__(4) LMNA carriers
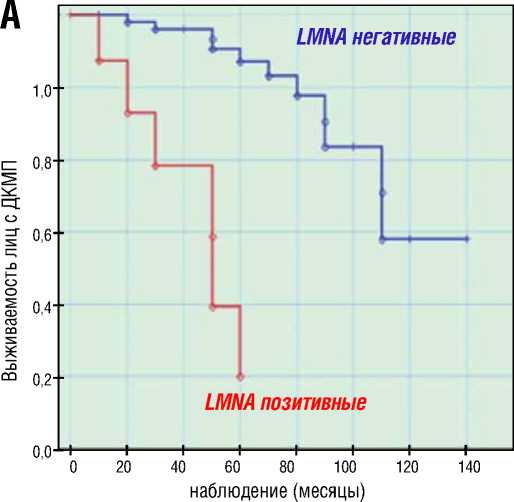
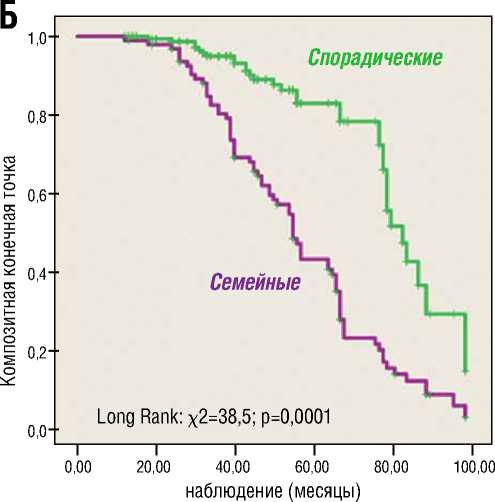
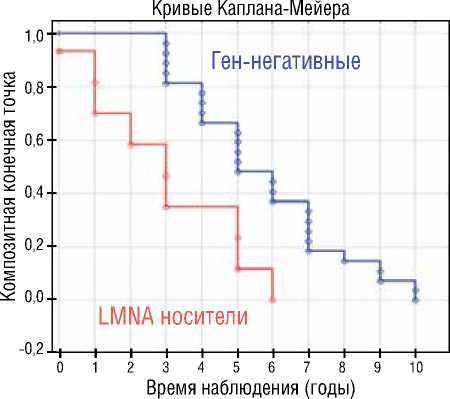
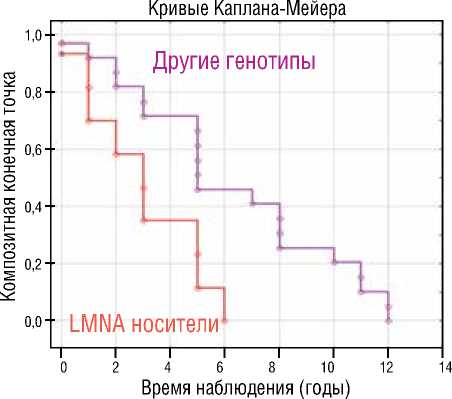
Рисунок 4. Кривые кумулятивной выживаемости Каплана-Мейера: графики дожития носителей LMNA мутаций vs LMNA негативных пациентов (А) и графики свободы от достижения композитной конечной точки (Б) пациентами с семейной и спорадической ДКМП
Figure 4. Kaplan-Meier cumulative survival curves: survival plots of LMNA mutation carriers vs LMNA negative patients (A) and freedom from reaching the composite endpoint (B) in patients with familial and sporadic DCM
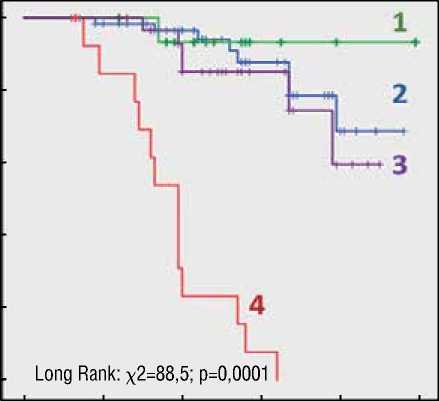
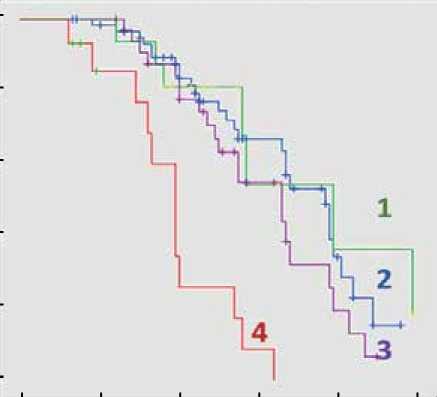
Рисунок 3. Кривые 5-летней бессобытийной выживаемости Каплана-Мейера: графики свободы от достижения первичной конечной точки (А) и композитной конечной точки (Б) в зависимости от генотипа ДКМП
Figure 3. Kaplan-Meier 5-year event-free survival curves: plots of freedom from reaching the primary endpoint (A)
and composite endpoint (B) according to DCM genotype
стигали пКТ и в два раза – кКТ (RR 2,13; 95% ДИ 1,51–3,01) по сравнению с носителями других генотипов. Риск летальности (RR 12,2; 95% ДИ 2,78–53,8; чувствительность 75%, специфичность 88%) у лиц c LMNA ассоциированной ДКМП в 12 раз превысил риск летального исхода у носителей других генотипов. Таким образом, вероятность развития всех неблагоприятных клинических исходов у LMNA -позитивных пациентов многократно превысила риск достижения пКТ и кКТ у LMNA -негативных пациентов: относительный риск развития жизнеопасной ЖТА/ВСС выше в 7,3 раз и риск кардиоваскулярной смерти выше в 6,6 раз; параметры относительного риска RR для разных генотипов ДКМП представлены в таблице 5.
Для сравнения генотипов и оценки влияния генетической составляющей на функцию жизни и достижение первичной и композитной конечных точек проведен анализ Каплана-Мейера с построением кривых выживаемости. Для оценки кривых применили лог-ранговый критерий (Mantel-Cox).
В результате анализа log-функций и долей в достижении первичной конечной точки, выявлена драматически низкая кумулятивная бессобытийная выживаемость пациентов с LMNA генотипом – самая низкая долевая свобода от ЖТА событий (пКТ) в период наблюдения ( χ 2=88,5; р=0,0001) по сравнению с другими генетическими группами (рис. 3А) и самый низкий уровень кумулятивной свободы от достижения кКТ (рис. 3Б) по сравнению с TTNtv генотипом ( χ 2=11,4; р=0,001), другими генотипами ( χ 2=15,4; р=0,0001) и ген-негативными пациентами с ДКМП ( χ 2=27,8; р=0,0001). Кривые выживаемости Каплана-Мейера представлены на рисунке 3.
Процедуру Каплана-Мейера с построением кривых выживаемости применили также для сравнения клинических исходов семейной, LMNA ассоциированной и спорадической формы ДКМП. В результате анализа log-функций и долей (определение доли пациентов с ДКМП, свободных от достижения кКТ), выявлены значимые прогностические различия между семейной и спорадической ДКМП, а также значительные отличия в кумулятивной выживаемости (доля свободы от летальности) между LMNA позитивными и LMNA негативными пациентами. Худший прогноз заболевания с высоким уровнем летальных исходов от всех кардиальных причин (кумулятивная выживаемость 20%) за 5 лет наблюдения продемонстрировали носители LMNA генотипа, в то время как бессобытийная выживаемость LMNA негативных пациентов составила 86% (рис. 4А). Значимо худший прогноз и низкий уровень бессобытийной выживаемости ( χ 2=38,5; р=0,0001) показали пациенты с семейной формой ДКМП (рис. 4Б). Достижение кКТ наблюдалось у 93,2% пробандов с семейной формой ДКМП, в группе спорадической ДКМП 45,8% лиц достигли комбинированной конечной точки.
Для оценки факторов риска неблагоприятных кардиоваскулярных событий (кКТ: летальный исход, ТС и уЖТ/ФЖ) применен пошаговый метод бинарной логистической регрессии. В результате логит-регрессионного анализа ( χ 2=30,3; p=0,00001) определены пять независимых предикторов риска неблагоприятных исходов ДКМП (достижение кКТ) – наличие мутации в гене LMNA (дихотомический фактор 0/1: b= - 2,1), диастолической дисфункции (Е/е’: b= - 0,01), митральной регургитации (МР, степень: b= - 1,05), АВ блокады (дихотомический фактор 0/1: b=0,25) и снижение глобальной продольной сократимости ЛЖ (GLS,%: b=0,06).
В математической модели коэффициенты регрессии продемонстрировали уровень значимости p <0,05 для всех 5-и предикторов риска и переменной- константы; максимальный коэффициент логистической регрессии определён для фактора LMNA позитивности (b= - 2,1; р=0,0002). Математическая формула логистической регрессии для калькулятора риска и обобщающий алгоритм клинической оценки в персонифицированной стратификации 5-летнего риска представлены на итоговом рисунке 5.
Для анализа качества построенной модели проведена оценка параметра отношения несогласия и ROC-анализ. Показатель отношения несогласия в представленной модели регрессии составил 5,6; значение логарифмического правдоподобия – 122. Результаты оценки качества прогностической модели по данным ROC анализа с построением характеристических кривых также подтвердили предиктивную корректность логит-модели (площадь под ROC-кривой AUC=0,765; 95% ДИ 0,699–0,897; р=0,0001; чувствительность 75%, специфичность 78%).
Таким образом, результаты проведенного исследования свидетельствуют о стратегической важности молекулярно-генетической диагностики как неотъемлемой части интегральной клинической оценки ДКМП для стратификации риска и определения прогноза. Представленные данные с убедительной доказательной силой демонстрируют крайнюю агрессивность клинического течения LMNA ассоциированных форм ДКМП, которые имеют (по сравнению с другими генотипами и генотип-негативными вариантами) более высокую частоту потенциально фатальных ЖТА, терминальной стадии СН и летальных исходов.
ОБСУЖДЕНИЕ
В представленной когорте патогенные варианты в гене LMNA обнаружены у 8,33% пациентов с ДКМП, эти результаты соответствует европейским литературным данным и «генетическому атласу» с уровнем распространенности до 5–8% [2]. В целом, мутации в гене LMNA (MIM 150330) составляют 0,5–5% случаев ДКМП, однако уровень диагностики ламинопатий повышается до 9–11% в когортах семейной ДКМП, а в семейных
Таблица 5. Относительный риск LMNA-позитивности: влияние LMNA-генотипа на клинические исходы ДКМП
Table 5. Relative risk of LMNA positivity: the impact of LMNA genotype on the clinical outcomes of DCM
|
Клинический исход ДКМП |
Относительный риск LMNA генотипа в сравнении с LMNA-негативным генотипом |
Относительный риск LMNA генотипа в сравнении с другими генотипами ДКМП |
Относительный риск LMNA генотипа в сравнении с генотип-негативной ДКМП |
|||
|
ОР (RR) |
95% ДИ |
ОР (RR) |
95% ДИ |
ОР (RR) |
95% ДИ |
|
|
Достижение пКТ |
7,33 |
4,63–11,6 |
6,12 |
3,17–11,8 |
8,31 |
4,54–13,2 |
|
Достижение кКТ |
2,36 |
1,82–3,09 |
2,13 |
1,51–3,01 |
2,52 |
1,84–3,45 |
|
Летальный исход |
6,60 |
2,86–15,3 |
12,2 |
2,78–53,8 |
5,19 |
2,14–12,6 |
Примечание: ОР – относительный риск, ДИ – доверительный интервал, пКТ – первичная конечная точка, кКТ – комбинированная
конечная точка
Note: RR – relative risk, CI – confidence interval, pEP – primary endpoint, cEP – composite endpoint
EURASIAN HEART JOURNAL, 2, 2023 35
случаях, ассоциированных с нарушениями АВ проводимости достигает 30–62% [3, 6, 7, 10, 13].
В представленной нами когорте варианты в гене LMNA были наиболее частой генетической причиной семейной ДКМП (11/21,6% из 51 PV/LPV). Следует отметить, что у 46,2% LMNA-носителей, перенесших уЖТ/ФЖ или полную АВ блокаду в качестве первого проявления болезни, не было выявлено значительной дилатации ЛЖ и значимой систолической дисфункции (ФВЛЖ не снижалась ниже 49% в течение нескольких лет). При последующем наблюдении эпизоды ЖТА наблюдались у 53,8% пациентов, а у 70% лиц с имплантированными девайсами регистрировали обоснованные ИКД разряды. Эти данные еще раз подтвердили высокую аритмогенность LMNA ассоциированной ДКМП и ограниченную прогностическую значимость систолической дисфункции как основного риск-стратифицирующего фактора (в пользу повышения стратегической важности генетического диагноза). Систолическая дисфункция, несомненно, является главным предиктором всех неблагоприятных исходов ДКМП, но для пациентов с ламино- патиями прогностическое значение низкой ФВЛЖ в оценке риска жизнеопасных ЖТА уступает показателю глобальной продольной сократимости и дефектам АВ проведения. У пациентов с LMNA ассоциированной ДКМП раннее появление интерстициального фиброза может быть причиной ЖТА задолго до развития систолической дисфункции. В исследованиях последних лет [14, 15] авторы сообщают о высокой распространенности МРТ признаков фиброза (с преимущественной локализацией в области межжелудочковой перегородки) у бессимптомных LMNA носителей; структурные изменения миокарда могут служить одним из потенциальных субстратов/ триггеров для желудочковых аритмий как первых клинических проявлений ДКМП. В представленном нами исследовании МРТ признаки фиброза выявлены у 92,3% пробандов и 31,3% родственников с LMNA ассоциированной ДКМП; в группе лиц с семейной ДКМП фиброз выявлен в 37% случаев, в отличие от спорадической формы – 15,7% (χ2=8,18; р=0,005).
Несмотря на то, что различия в прогнозе между семейными и спорадическими формами ДКМП остаются в настоящее
СЕМЕЙНАЯ ФОРМА ДКМП N=73; 47%
СПОРАДИЧЕСКАЯ ФОРМА ДКМП N=83; 53%
|
Возраст дебюта ДКМП |
43 [34; 48] |
|
Возраст дебюта ФП |
38 [34; 49] |
|
Дебют АВ блокады |
33 [23; 44] |
Возраст ТС, лет
40 [37; 49]
Возраст дебюта ДКМП 33 [25;43]
Возраст дебюта ФП 35 [30; 41]
Дебют АВ блокады 31 [25; 36]
Возраст ТС, лет 35 [30; 40]
Генетическая причина ДКМП n=19 (22,9%)
Генетическая причина не выявлена n=64 (77,1%)
Генетическая причина ДКМП Генетическая причина n=47 (64,4%) не выявлена n=26 (35,6%)
ГЕНЕТИЧЕСКАЯ КЛАССИФИКАЦИЯ И ГЕНОТИП-СПЕЦИФИЧЕСКИЙ ПРОГНОЗ

LMNA
TTNtv
BAG3
RBM20
DSP
SCN5A
MYH7
Стратификация риска по формуле Р = 1/1+ e-Z согласно бинарной логит регрессии:
Z = 2,24 - 2,1 х LMNA (0/1) - 1,05 х MR (степень) + 0,06 х GLS(%) - 0,01 х E/e` + 0,25 х AV блокада (0/1)
Рисунок 5. Персонализированная модель классификации для оценки 5-летнего риска неблагоприятных исходов ДКМПFigure 5. Personalized classification for assessing the 5-year risk of adverse outcomes of DCM
36 ЕВРАЗИЙСКИЙ КАРДИОЛОГИЧЕСКИЙ ЖУРНАЛ, 2, 2023
время спорными [13–16], своевременная доклиническая диагностика наследственной ДКМП (обусловленной, в частности, аритмогенными LMNA, RBM20, PLN, FLNC мутациями) у потомства и родственников является крайне важным аспектом для принятия комплекса превентивных мер.
Результаты представленного нами исследования продемонстрировали худший прогноз и низкий уровень кумулятивной выживаемости пациентов с семейной ДКМП, а также драматический разрыв между рутинной диагностикой семейной формы ДКМП (7/4,5%) и возможностью многократно повысить уровень выявления серьезного семейного заболевания (до 73/46,8%) с помощью каскадного скрининга и генетического тестирования. Полученные нами результаты подтверждают выводы авторов G. Huggins et al. (2022) о существенной гиподиагностике семейной ДКМП в рутинной практике – «идиопатическая ДКМП гораздо чаще является семейной, чем это обычно устанавливается»; на основании семейного анамнеза только у ~5% пациентов с ДКМП можно заподозрить семейное заболевание, тогда как каскадный скрининг позволяет верифицировать семейную форму ДКМП, как минимум, в 29,7% случаев [16]. Обнаружение каузального генетического варианта у пробанда позволяет проводить прогностическое тестирование членов семьи и профилактическое вмешательство. Несмотря на то, что ген-специфическая терапия остается пока, в основном, в сфере высоко технологичных исследований, все же хорошо изученные к настоящему времени генотип-фенотипические корреляции позволяют персонализировать тактику и методы лечения. В первую очередь это относится к мониторингу носителей вариантов в генах LMNA, PLN, FLNC и RBM20 , у которых развиваются злокачественные аритмии (несмотря на относительно сохранную ФВЛЖ), – стандартные рекомендации для первичной ИКД-профилактики не подходят для этих пациентов [17]. Пациенты с LMNA и RBM20 вариантами также могут нуждаться в раннем рассмотрении необходимости проведения ТС, так как они часто имеют быстро прогрессирующее течение сердечной недостаточности [10, 13]. Для бессимптомных родственников, носителей этих вариантов, крайне важен мониторинг доклинической дисфункции желудочков с применением ЭхоКГ методов отслеживания (спекл-трекинг) и МРТ сердца с контрастированием [17].
ЗАКЛЮЧЕНИЕ
-
1. Каскадный семейный скрининг (клиническое обследование и генетическое тестирование) позволил повысить уровень диагностики семейных форм в когорте ДКМП с 4,5% до 46,8%.
-
2. У пациентов с семейной и спорадической ДКМП обнаружено сходство генетического спектра. Однако LMNA варианты были наиболее частой причиной семейной ДКМП, а TTNtv доминировали у лиц со спорадической ДКМП (ОШ 6,19; 95% ДИ 1,04–36,8).
-
3. Пациенты с семейной ДКМП показали худший 5-летний прогноз и низкую кумулятивную бессобытийную выживаемость в сравнении с группой спорадической ДКМП (log rank χ 2=38,5; р=0,0001).
-
4. Результаты проведенного исследования подтвердили высокую аритмогенность LMNA генотипа, выявлены высоко значимые прогностические ассоциации ламинопа-тий с эпизодами жизнеопасных ЖТА ( χ 2=39,9; р=0,0001).
-
5. В результате логит-регрессионного анализа определены пять независимых предикторов ( χ 2=30,3; p=0,0001) 5-летнего риска развития неблагоприятных исходов ДКМП – наличие мутации в гене LMNA , диастолической дисфункции (Е/е’), выраженной митральной регургита-
- ции и АВ блокады, а также снижение глобальной продольной сократимости ЛЖ (GLS).
Список литературы Генетический спектр семейной и спорадической дилатационной кардиомиопатии: аритмические фенотипы, ассоциированные с мутациями в гене ламина A/C (LMNA)
- Merlo M., Cannatà A., Gobbo M. et al. Evolving concepts in dilated cardiomyopathy. Eur J Heart Fail. 2018;20(2):228–339. https://doi.org/10.1002/ejhf.1103
- Paldino A., De Angelis G., Merlo M. et al. Genetics of dilated cardiomyopathy: clinical implications. Curr Cardiol Rep. 2018;20(10):83. https://doi.org/10.1007/s11886-018-1030-7
- Hershberger R.E., Cowan J., Jordan E. et al. The complex and diverse genetic architecture of dilated cardiomyopathy. Circ Res. 2021;128:1514–1532. https://doi.org/10.1161/circresaha.121.318157
- Jordan E., Peterson L., Ai T. et al. Evidence-Based Assessment of Genes in Dilated Cardiomyopathy.Circulation.2021;144(1):7-19. https://doi.org/10.1161/circulationaha.120.053033
- Akhtar M.M., Lorenzini M., Cicerchia M. et al. Clinical phenotypes and prognosis of dilated cardiomyopathy caused by truncating variants in the TTN Gene. Circ Heart Fail. 2020;13(10):e006832. https://doi.org/10.1161/circheartfailure.119.006832
- Hasselberg N.E., Haland T.F., Saberniak J. et al. Lamin A/C cardiomyopathy: young onset, high penetrance, and frequent need for heart transplantation. Eur Heart J. 2018;39(10):853–860. https://doi.org/10.1093/eurheartj/ehx596
- Captur G., Arbustini E., Bonne G. et al. Lamin and the heart. Heart. 2018;104(6):468–479. http://dx.doi.org/10.1136/heartjnl-2017-312338
- Marian A.J. Clinical interpretation and management of genetic variants. JACC Basic Transl Sci. 2020;5:1029–1042. https://doi.org/10.1016/j.jacbts.2020.05.013
- Richards S., Aziz N., Bale S. et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405–424. https://doi.org/10.1038/gim.2015.30
- Peters S., Johnson R., Fatkin D. et al. Familial Dilated Cardiomyopathy. Heart Lung Circ. 2020;29(4):566-574. https://doi.org/10.1016/j.hlc.2019.11.018
- Priori S.G., Blomström-Lundqvist C., Mazzanti A. et al. ESC Scientific Document Group. 2015 ESC guidelines for the management of patients with ventricular arrhythmias and the prevention of sudden cardiac death: the task force for the management of patients with ventricular arrhythmias and the prevention of sudden cardiac death of the European Society of Cardiology (ESC). endorsed by: association for European Paediatric and Congenital Cardiology (AEPC). Eur Heart J. 2015;36:2793–2867. https://doi.org/10.1093/eurheartj/ehv316
- Wahbi K., Ben Yaou R., Gandjbakhch E. et al. Development and Validation of a New Risk Prediction Score for Life-Threatening Ventricular Tachyarrhythmias in Laminopathies. Circulation. 2019;140(4):293-302. https://doi.org/10.1161/circulationaha.118.039410
- Gigli M., Merlo M., Graw S.L. et al. Genetic risk of arrhythmic phenotypes in patients with dilated cardiomyopathy. J Am Coll Cardiol. 2019;74(11):1480–1490. https://doi.org/10.1016/j.jacc.2019.06.072
- Peretto G., Barison A., Forleo C. et al. Late gadolinium enhancement role in arrhythmic risk stratification of patients with LMNA cardiomyopathy: results from a long-term follow-up multicentre study. Europace 2020;22:1864–72. https://doi.org/10.1093/europace/euaa171
- Tayal U., Verdonschot J.A.J., Hazebroek M.R. et al. Precision phenotyping of dilated cardiomyopathy using multidimensional data. J Am Coll Cardiol.2022;79:2219–2232. https://doi.org/10.1016/j.jacc.2022.03.375
- Huggins G.S., Kinnamon D.D., Haas G.J. et al. DCM Precision Medicine Study of the DCM Consortium. Prevalence and cumulative risk of familial idiopathic dilated cardiomyopathy. JAMA. 2022;327(5):454–463. https://doi.org/10.1001/jama.2021.24674
- Zeppenfeld K., Tfelt-Hansen J., Marta de Riva. et al. 2022 ESC Guidelines for the management of patients with ventricular arrhythmias and the prevention of sudden cardiac death: Developed by the task force for the management of patients with ventricular arrhythmias and the prevention of sudden cardiac death of the European Society of Cardiology (ESC) Endorsed by the Association for European Paediatric and Congenital Cardiology (AEPC). European Heart Journal. 2022;43(40):3997–4126. https://doi.org/10.1093/eurheartj/ehac262
