Геномный ландшафт колоректального рака. Пилотное исследование в Российской Федерации

Автор: Семина Е.В., Кубасова К.А., Какоткин В.В., Родимов С.В., Агапов М.А.
Журнал: Сибирский онкологический журнал @siboncoj
Рубрика: Лабораторные и экспериментальные исследования
Статья в выпуске: 3 т.24, 2025 года.
Бесплатный доступ
Цель исследования – комплексный анализ молекулярно-генетических изменений в опухолевых и нормальных тканях больных колоректальным раком с использованием полногеномного секвенирования для выявления мутаций de novo, паттернов микросателлитной нестабильности и мутационных сигнатур, связанных с возникновением опухоли. Материал и методы. Исследование являлось одномоментным поперечным, проводилось на территории Калининградской области. Проведен анализ образцов колоректального рака с использованием полногеномного секвенирования нового поколения (WGS). Для картирования прочтений был использован референсный геном человека GRCh38.p14. Биоинформатический анализ сигнатур проводился отдельно по каждому пациенту. Была секвенирована 71 пара образцов: полные геномы биопсии опухоли и ДНК, выделенная из крови, для 71 пациента. Средний возраст пациентов – 69 лет, медианный – 71 год. Результаты. Выявлены генетические изменения, связанные с развитием и прогрессированием опухоли, а также потенциальные терапевтические мишени. Используя базу данных Oncovar, была обнаружена 151 драйверная мутация у 62 пациентов. С использованием базы данных CIVIC выявлено 70 маркерных мутаций у 45 пациентов. Обнаружено, что самые распространенные драйверные мутации в исследуемой когорте – мутации в гене KRAS (KRAS:G35T, KRAS:G35A, KRAS:G38A), ассоциированные с устойчивостью опухоли к препаратам Панитумумаб и Цетуксимаб. Частота наиболее распространенных драйверных мутаций в исследуемой когорте низкая, что говорит о высокой гетерогенности колоректального рака. При анализе мутационной нагрузки средний показатель TMB был равен 5,5, что свидетельствует о высоком потенциале использования иммунотерапии при колоректальном раке. В исследуемой популяции преобладают мутационные сигнатуры SBS1, SBS5 и SBS39. Заключение. Выявленное многообразие изменений генома может быть связано с ошибками в репликации ДНК, воздействием внешних факторов и генетической предрасположенностью. Профиль мутационных сигнатур очень схож в исследуемых образцах всех пациентов, что предполагает действие одного/единого мутагенного фактора в развитии колоректального рака у всех пациентов.
Колоректальный рак, полногеномное секвенирование, секвенирование нового поколения, Next-Generation Sequencing, NGS
Короткий адрес: https://sciup.org/140310575
IDR: 140310575 | УДК: 616.345+616.351]-006.6 (470+571): 577.323.432 | DOI: 10.21294/1814-4861-2025-24-3-65-75
Genetic landscape of colorectal cancer. A pilot study in the Russian Federation
The purpose of the study was a comprehensive analysis of molecular genetic changes in tumor and normal tissues of patients with colorectal cancer using whole-genome sequencing to identify de novo mutations, microsatellite instability patterns, and mutational signatures associated with tumor development. Material and Methods. A single-time cross-sectional study was conducted in the Kaliningrad region. Colorectal cancer samples were analyzed using whole-genome next-generation sequencing (WGS). The human reference genome GRCh38.p14 was used for read mapping. Bioinformatic analysis of signatures was conducted separately for each patient. 71 pairs of samples were sequenced: complete genomes of tumor biopsies and DNA isolated from blood samples for 71 patients. The average age among the analyzed patients was 69 years, with a median of 71 years. Results. Genetic alterations associated with tumor development and progression, as well as potential therapeutic targets were identified. Using the Oncovar database, 151 driver mutations were detected in 62 patients. Using the CIVIC database, 70 marker mutations were found in 45 patients. It was found that the most common driver mutations in the study cohort were mutations in the KRAS gene (KRAS:G35T, KRAS:G35A, KRAS:G38A), associated with tumor resistance to Panitumumab and Cetuximab. The frequency of the most common driver mutations in the study cohort was low, indicating high heterogeneity of colorectal cancer. Analysis of the tumor mutational burden (TMB) revealed a high average value of 5.5, suggesting a significant potential of immunotherapy in colorectal cancer treatment. In the studied population, the mutational signatures SBS1, SBS5, and SBS39 were the most common. Conclusion. The identified diversity of genomic changes may be associated with DNA replication errors, exposure to external factors, and genetic predisposition. The pattern of mutational signatures was very similar among all patients in the studied samples, suggesting a single mutagenic factor is involved in the development of colorectal cancer in all patients.
Текст научной статьи Геномный ландшафт колоректального рака. Пилотное исследование в Российской Федерации
Колоректальный рак (КРР) является одной из наиболее распространенных и агрессивных форм злокачественных новообразований, он входит в число основных причин смертности от онкологических заболеваний по всему миру. Понимание молекулярной природы этого заболевания имеет критическое значение для разработки методов диагностики, прогноза и терапии. В последние годы значительно расширились знания о генетических и эпигенетических изменениях, ассоциированных с развитием и прогрессированием КРР. Одним из ключевых методов исследования геномных изменений в образцах КРР является секвенирование нового поколения (Next-Generation Sequencing, NGS), которое позволяет проводить глубокий анализ геномных и транскриптомных изменений с высокой степенью чувствительности и точности.
NGS предоставляет возможности для выявления точечных мутаций, копийных вариаций, а также изменений в структуре генома, что особенно важно для изучения гетерогенности опухолей и индивидуальных особенностей патогенеза КРР у различных пациентов. В частности, значительное внимание уделяется детекции соматических мутаций, т. к. они могут служить потенциальными биомаркерами для ранней диагностики, прогноза и предсказания ответа на терапию. Генетические изменения в таких ключевых генах, как APC, KRAS, TP53, PIK3CA, а также в участках микросателлит-ной нестабильности (MSI) могут существенно повлиять на прогноз заболевания и ответ на лечение. Современные NGS-технологии, такие как панели таргетного секвенирования, полногеномное и эк-зомное секвенирование, открывают возможность для создания индивидуализированных подходов к лечению пациентов с КРР.
Обработка данных NGS представляет собой многосоставной процесс, включающий этапы выравнивания последовательностей на референсный геном, выявления и аннотации вариантов, фильтрации артефактов и ложноположительных результатов, а также интерпретации биологически значимых изменений. Точность анализа NGS данных играет решающую роль в получении достоверных результатов, которые могут быть применены в клинической практике.
В отечественной литературе описаны пилотные исследования, направленные на идентификацию наследственных форм онкологических заболеваний у лиц с повышенным риском развития и использование образцов крови для этой цели [1]. Однако на российской выборке больных отсутствуют исследования, включающие анализ первичного опухолевого узла и образца крови как контроля мутаций и исключающие из анализа мутации, обнаруженные в крови, для выявления мутаций de novo , которые могут быть связаны с возникновением опухоли.
В настоящем исследовании впервые на российской популяции проведен анализ 71 образца КРР с использованием полногеномного NGS (WGS). В работе представлен алгоритм обработки данных NGS и анализа мутаций, выявленных в образцах колоректального рака. Описание этапов обработки данных включает подготовку первичного (сырого) материала, фильтрацию и аннотирование полученных данных, а также их интерпретацию.
Цель исследования – комплексный анализ молекулярно-генетических изменений в опухолевых и нормальных тканях больных колоректальным раком с использованием полногеномного секвенирования для выявления мутаций de novo , паттернов микросателлитной нестабильности и мутационных сигнатур, связанных с возникновением опухоли.
Материал и методы
Исследование являлось одномоментным поперечным, проводилось на территории Калининградской области в рамках проекта «Национальная генетическая инициатива 100000+Я». Критерии включения в исследование: возраст старше 18 лет; подписанное информированное согласие на участие в исследовании и использование биологического материала в целях исследования; обнаружение при колоноскопии эпителиального образования (типы 2B-3 по JNET, наличие ямочного рисунка Vi и Vn по S. Kudo) с последующей верификацией диагноза «аденокарцинома толстой кишки» или «аденома толстой кишки» с помощью морфологического исследования.
Критерии невключения: возраст до 18 лет; наличие носительства вирусных инфекций с парентеральным путем передачи (ВИЧ, гепатит С, гепатит В, сифилис); отказ от участия в исследовании или невозможность получить согласие из-за мнестико-интеллектуальных нарушений или иных причин; невозможность получения достаточного объема биологического материала (фрагмента слизистой оболочки кишки) для проведения рутинного патологоанатомического исследования и выделения ДНК.
Критерии исключения: отсутствие опухолевого роста в полученных фрагментах слизистой по результатам морфологического исследования; отказ добровольца от обработки результатов секвенирования ДНК, полученной из биологического материала, на любом этапе.
Для участия в исследовании приглашались пациенты, обратившиеся для выполнения эндоскопического скрининга колоректального рака. Непосредственно на этапе подготовки к процедуре пациенты были информированы о цели, задачах исследования, возможности участия в исследовании в качестве добровольца в случае выявления по данным первичного эндоскопического скрининга новообразований ободочной кишки, характеризующихся наличием паттернов метаплазии, дисплазии или злокачественной трансформации. В случае подписания пациентом добровольного информированного согласия и обнаружения образования в толстой кишке производился забор фрагмента слизистой оболочки толстой кишки в наиболее измененной части, объемом 2–3 мм3, который помещался в пробирку с буферным раствором, а также 16 мл венозной крови.
В случае выявления критериев невключения/ исключения на этапе эндоскопического или морфологического исследования врач-исследователь сообщал об этом добровольцам, а биологические образцы подлежали утилизации.
Геномная ДНК была выделена методом сорбции на магнитных частицах (набор MGIEasy Magnetic Beads Blood Genomic DNA Extraction Kit, MGI), оценка качества выделенной геномной ДНК проведена методами спектрофотометрии и флюорометрии, затем использована для приготовления геномных библиотек для секвенирования (набор для подготовки библиотек без ПЦР, с энзиматической фрагментацией ДНК MGIEasy FS PCR-Free Library Prep Set, 96 реакций (MIX), MGI). Полученные библиотеки анализировали на секвенаторе (DNBSEQ-T7), методом PE150.
Для тримминга и фильтрации прочтений был использован инструмент fastp [2, OpenGene/fastp] со следующими параметрами: -5 -3 -W 4 -M 20 -l 70 -q 20 -u 20. Тримминг производился с 5’ и с 3’ конца прочтения (-5, -3) плавающим окном длиной 4 нуклеотида (-W 4). Если среднее качество прочтений внутри плавающего окна было меньше 30 (-M 30), то этот фрагмент обрезался. Также отфильтровывались прочтения с длиной после тримминга меньше 70 (-l 70), а также прочтения, у которых после тримминга более 30 % нуклеотидов имели качество ниже Q30 (-q 30, -u 30). Последовательности адаптеров детектировались и удалялись автоматически.
Для картирования прочтений был использован референсный геном человека GRCh38.p14, скачанный из Ensembl Homo_sapiens/Info/Index). Картирование про- чтений на индексированный геном выполнялось при помощи программы bwa mem со следующими параметрами: -M -v 3 [3]. Для маркирования дуплицированных прочтений была использована программа Picard MarkDuplicates с опцией -MAX_ RECORDS_IN_RAM 50000000. Прочтения с маркированными дупликатами в формате bam были проиндексированы при помощи samtools index.
Для поиска герминальных мутаций (SNP, InDel) использована программа DeepVariant [4, ]. DeepVariant был запущен в контейнере Singularity (google/ deepvariant:latest) с параметром --model_ type=»WGS» и остальными настройками по умолчанию. Для поиска соматических мутаций (SNP, InDel) был использован инструмент Strelka2 со стандартными настройками [5, Illumina/strelka], поскольку этот инструмент продемонстрировал высокую точность при выявлении соматических мутаций в ряде сравнительных исследований [6]. Strelka2 работает гораздо быстрее, что важно при обработке большого количества тяжелых данных полногеномного секвенирования. Ансамбли из инструментов незначительно повышают чувствительность или специфичность, но значительно увеличивают вычислительную нагрузку на анализ образцов. Фильтр в виде частоты мутации был использован, т. к. показана низкая точность определения мутаций с частотой меньше 5 %, особенно при секвенировании с глубиной 100X.
Для аннотации мутаций использована программа Ensembl VEP [7, info/docs/tools/vep/script/]. Для этой программы были использованы следующие параметры: «--sift b --polyphen b --protein –symbol --ccds --uniprot --per_gene --domains --check_existing --clin_sig_allele 1 --max_af--af_1kg --af_gnomade --pubmed --var_synonyms --failed 1». Также по каждой мутации был произведен поиск в базе данных CIVIC [, база данных Evidence] и в базе данных Oncovar [].
Для поиска амплификаций и делеций (вариантов числа копий – CNV) использована программа CNVkit [8, ]. Данная программа запущена в режиме batch (cnvkit. py batch), с опциями -m wgs, –annotate , –target-avg-size 1000. Для аннотации найденных CNV и определения количества копий каждого гена использована аннотация генома человека версии GRCh38, скачанная с сайта USCS ).
Для оценки качества сырых прочтений в формате fastq использована программа FASTQC [9, fastqc/]. Графики распределения качества, ГЦ-состава и неопознанных нуклеотидов (N) по позиции в прочтении после тримминга и фильтрации получены при помощи программы fastp [2, ]. Анализ контаминации в образце выполнен при помощи программы FastQ_Screen [10, https://www.bioinformatics. ]. Для анализа качества картирования прочтений на референсный геном использована программа Picard, функции CollectAlignmentSummaryMetrics и CollectInsertSizeMetrics [ io/picard/]. Получены график доли картированных на геном прочтений и график распределения размера вставки (insert size). Для анализа покрытия референсного генома использована программа Mosdepth [11, ].
Для анализа микросателлитной нестабильности (MSI) использован инструмент MSISencor-Pro [12, ]. Для запуска этой программы на первом этапе подготовлен файл с микросателлитами генома человека, используя команду msisensor-pro scan и референсный геном (GRCh38.p14) в формате fasta. Затем, используя этот файл и файлы с картированием на геном прочтений нормы и опухоли (.bam файлы), выполнен запуск программы msisensor-pro msi со стандартными настройками. В результате получен файл в формате tsv, который содержал следующую информацию: количество сайтов с микросателлитами в геноме человека, количество сайтов с нестабильными микросателлитами в образце и доля нестабильных микросателлитных сайтов (MSI) в образце.
Для анализа дефектов гомологичной рекомбинации (HRD) в образце использована программа HRDCNA, выдающая значения в диапазоне от 0 до 1 [13, 2023, ]. Для запуска этой программы использован файл с количеством копий сегментов (*, полученный при помощи программы CNVkit. В результате работы программы получен HRDCNAScore для образца (значение от 0 до 1). Чем выше значение HRDCNAScore, тем выше вероятность того, что в образце присутствуют дефекты гомологичной рекомбинации.
Биоинформатический анализ сигнатур проводился отдельно по каждому пациенту. Для анализа мутационных сигнатур использована программа SigProfilerAssignment [14, https://github. com/ShixiangWang/sigminer/]. Для запуска этой программы использован vcf файл с соматическими мутациями (SNP, InDel). На выходе получена таблица со значением вклада каждой мутационной сигнатуры (COSMIC v.3.4, в формирование мутаций у пациента.
Результаты
В данной работе секвенирована 71 пара образцов: полные геномы биопсии опухоли и ДНК, выделенная из крови, для 71 пациента. Средний
Таблица/Table
Клинико-патоморфологические характеристики больных КРР
Clinical and pathological characteristics of patients with colorectal cancer (CRC)
|
Признак/Sing |
Доля в выборке/Proportion |
|
Локализация опухоли/Tumor localization |
|
|
Прямая кишка/Rectum |
17 (23,94 %) |
|
Ректосигмоидный переход/Rectosigmoid junction |
8 (11,27 %) |
|
Сигмовидная кишка/Sigmoid colon |
18 (25,35 %) |
|
Вышележащие отделы/Upper colon |
28 (39,44 %) |
|
Степень злокачественности/Tumor grade |
|
|
High grade |
64 (90,14 %) |
|
Low grade |
7 (9,86 %) |
|
Стадия опухолевого процесса/Tumor stage |
|
|
I |
9 (12,68 %) |
|
II |
18 (25,36 %) |
|
III |
21 (29,57 %) |
|
IV |
23 (32,39 %) |
|
pT |
|
|
pT2 |
11 (15,49 %) |
|
pT3 |
41 (57,75 %) |
|
pT4a |
15 (21,13 %) |
|
pT4b |
4 (5,63 %) |
|
pN |
|
|
pN0 |
28 (39,44 %) |
|
pN1a |
22 (30,99 %) |
|
pN1b |
5 (7,04 %) |
|
pN1c |
3 (4,23 %) |
|
pN2a |
5 (7,04 %) |
|
pN2b |
8 (11,26 %) |
|
M |
|
|
cM0 |
48 (67,61 %) |
|
pM1a |
12 (16,90 %) |
|
pM1b |
11 (15,49 %) |
|
Лимфатическая инвазия/Lymphovascular invasion |
|
|
L0 |
43 (60,56 %) |
|
L1 |
28 (39,44 %) |
|
Сосудистая инвазия/Vascular invasion |
|
|
V0 |
62 (87,32 %) |
|
V1 |
9 (12,68 %) |
|
Периневральная инвазия/Perineural invasion |
|
|
Pn0 |
60 (84,51 %) |
|
Pn1 |
11 (15,49 %) |
|
Опухолевый баддинг/Tumor budding |
|
|
Bd1 |
32 (45,07 %) |
|
Bd2 |
26 (36,62 %) |
|
Bd3 |
13 (18,31 %) |
Примечание: таблица составлена авторами.
Note: created by the authors.
возраст пациентов – 69 (SD=11,1) лет, медианный – 71 (Q1/Q3=61/75) год (рис. 1). Распределение по полу: 35 мужчин, 36 женщин (рис. 1). Клиникопатоморфологические характеристики больных КРР представлены в таблице.
После фильтрации прочтений средний процент оснований с качеством не менее Q30 в образцах нормы равняется 92,3 %, в образцах опухоли – 91,9 %.
На референсный геном было выровнено 100 % прочтений нормы и 99,4 % прочтений опухоли. Средний процент дуплицированных прочтений в образцах нормы равняется 4,4 %, в образцах опухоли – 13,2 %. Средний размер вставки в образцах нормы – 388,6, в образцах опухоли – 395,5. Средняя глубина прочтения образцов нормы – 26.9Х, образов опухоли – 100.0Х.
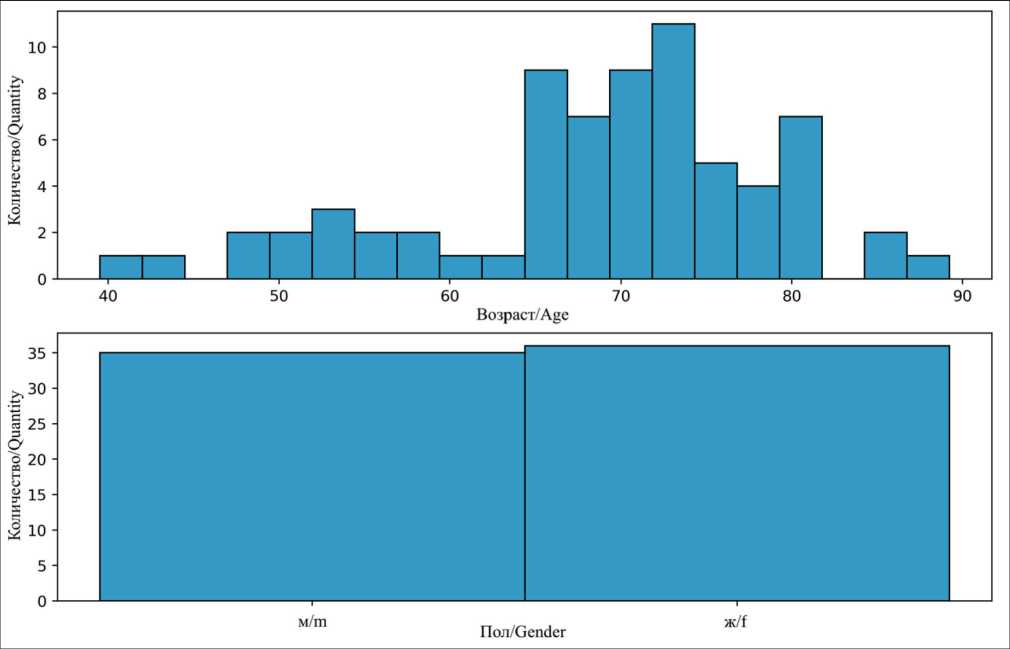
Рис. 1. Распределение пациентов по возрасту и полу. Горизонтальная ось – значение признака (возраст на верхнем графике и пол на нижнем графике), вертикальная ось – количество пациентов с данным значением признака; рисунок выполнен авторами Fig. 1. Patients’ distribution charts by age and gender. The horizontal axis is the characteristic value (age on the top graph and gender on the bottom graph), the vertical axis is the number of patients with given characteristic value; created by the authors
Среднее количество обнаруженных сырых соматических мутаций до фильтрации в одном образце – 1462120. После фильтрации соматических мутаций (были оставлены только мутации, влияющие на белок, с частотой мутантного аллеля >5 % и с количеством прочтений, содержащих мутантный аллель, не менее 5) в среднем для каждого пациента осталось 328 мутаций. Всего после такой фильтрации в выборке обнаружено 22 275 соматических мутаций. Больше всего мутаций было обнаружено в генах APC (79 мутаций у 50 пациентов), TTN (63 мутации у 32 пациентов), TP53 (50 мутаций у 46 пациентов), MUC19 (42 мутации у 17 пациентов), MUC4 (36 мутаций у 24 пациентов), KRAS (32 мутации у 30 пациентов).
Наиболее представленными конкретными мутациями среди обнаруженных генов были следующие: ENST00000311936(KRAS):c.225C>A (у 8 пациентов), ENST00000311936(KRAS):c.228C>T (у 6 пациентов), ENST00000311936(KRAS):c.225C>T (у 6 пациентов) и ENST00000634670(KLF18):c.2224-2225A>TAT (у 7 пациентов). Таблица «Top_ somatic_mutations» со всеми уникальными мутациями, сортированными по убыванию частоты встречаемости, приведена в приложении электронного варианта статьи.
Далее, используя базу данных Oncovar, нами обнаружена 151 драйверная мутация у 62 пациентов. Самые частые драйверные мутации – KRAS: G35T (8 пациентов), KRAS:G35A (6 пациентов), KRAS:G38A (6 пациентов). Таблица «Top_driver_ mutations» со всеми уникальными онкогенными мутациями, сортированными по убыванию частоты встречаемости, приведена в приложении электронного варианта статьи. Используя базу данных CIVIC, обнаружено 70 маркерных мутаций у 45 пациентов. Наиболее частые из них: KRAS:G35T (8 пациентов), KRAS:G35A (6 пациентов), KRAS:G38A (6 пациентов). Также у 4 пациентов идентифицирована маркерная мутация в гене BRAF V600E. Таблица «Top_marker_mutations» со всеми уникальными маркерными мутациями, сортированными по убыванию частоты встречаемости, приведена в приложении электронного варианта статьи.
Среднее количество обнаруженных герминальных мутаций в образце – 5 694 810. После фильтрации герминальных мутаций были оставлены только мутации, влияющие на белок в 80 генах (полный перечень приведен в Приложении), их оказалось 4 713. Больше всего мутаций было обнаружено в генах HLA-B (2 450 мутаций у 67 пациентов), EIF3E (227 мутаций у 60 пациентов), PMS2 (194 мутации у 66 пациентов), RNF43 (131 мутация у 65 пациентов), TP53 (129 мутаций у 66 пациентов). Таблица «Top_germline_mutations» со всеми уникальными герминальными мутациями в 80 генах, сортированными по убыванию частоты встречаемости, приведена в приложении электронного варианта статьи.
В среднем у каждого пациента обнаружены амплификации 2 554 генов и делеции 2 619 генов. Среди онкогенов по версии базы данных Oncovar наиболее часто встречаются амплификации в следующих генах (в скобках указано число образцов, имеющих мутацию): GNAS (41), KCNQ2 (41), ACTB (28), COL4A2 (29), SOSTDC1 (27), FERD3L (27),
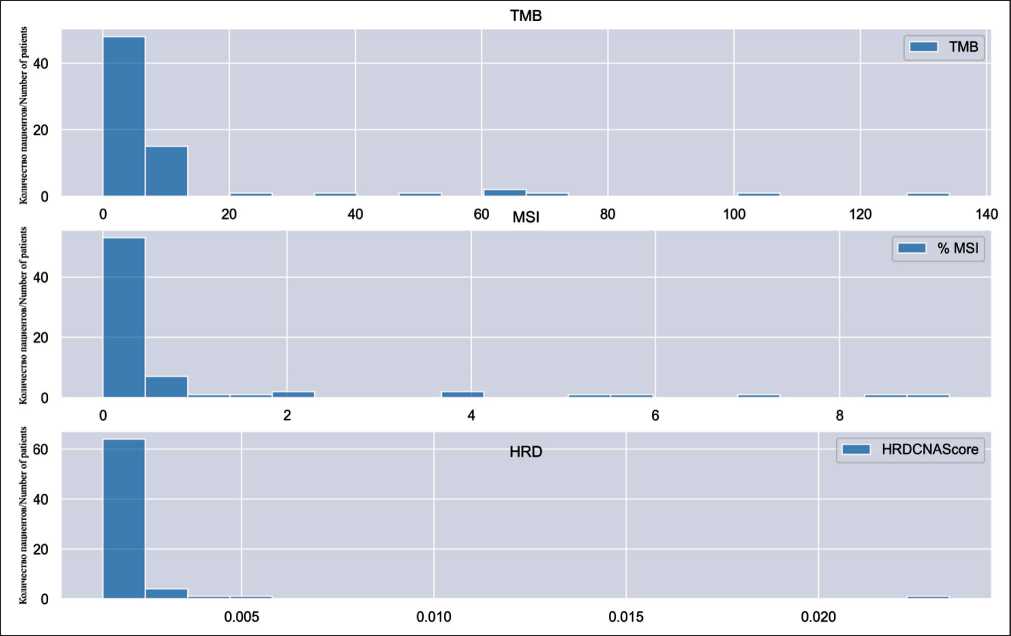
Рис. 2. Распределение TMB (мутационная нагрузка), MSI (доля сайтов с микросателлитной нестабильностью) и HRD (вероятность наличия дефектов в системе репарации путем гомологичной рекомбинации). Горизонтальная ось – значение параметра (TMB на верхнем графике, MSI на среднем и HRD на нижнем), вертикальная ось – количество пациентов с данным значением параметра; рисунок выполнен авторами
Fig. 2. Distribution charts of TMB (tumor mutational burden), MSI (proportion of sites with microsatellite instability) and HRD (defects’ probability in the repair system by homologous recombination). The horizontal axis represents the parameter value (TMB on the top chart, MSI on the middle chart and HRD on the bottom chart), the vertical axis represents the number of patients with a given parameter value; created by the authors
BRAF (23), ADCY8 (24), TRPS1 (23). Делеции наиболее часто встречаются в генах HDX (38), NKAP (38), THOC2 (38), AFF2 (38), RS1 (36), TMX3 (32), SMAD4 (32), LIPG (31), TP53 (23). Таблица «Top_ amplifications_deletions» (страница «Амплификации») со всеми амплификациями, сортированными по убыванию частоты встречаемости, приведена в приложении электронного варианта статьи. Таблица «Top_amplifications_deletions» (страница «Делеции») со всеми делециями, сортированными по убыванию частоты встречаемости, приведена в приложении электронного варианта статьи.
Согласно руководству FDA [15], высокий уровень TMB определяется по пороговому значению больше 10. Среднее значение TMB (количество мутаций на 1 млн пар оснований в геноме) составило 12,8 (SD=23,61, коэффициент вариации – 185,14 %), медианное значение – 5,5 (Q1/Q3=4,16/8,32) (рис. 2). Среднее значение MSI (процент сайтов с микро-сателлитной нестабильностью) составило 0,73 (SD=1,75, коэффициент вариации – 240,85 %), медианное значение – 0,07 (Q1/Q3=0,0/0,41) (рис. 2). Среднее значение HRD (значение, отображающее вероятность наличия дефектов в системе репарации путем гомологичной рекомбинации) составило 0,0019 (SD=0,0027, коэффициент вариации – 145,42 %), медианное значение – 0,0014 (Q1/ Q3=0,0013/0,0014) (рис. 2). Максимальное значение HRD – 0,0234. Такие низкие значения HRD могут свидетельствовать о том, что в исследуемой когор- те больных отсутствовали пациенты со значимыми дефектами в системе репарации двуцепочечных разрывов ДНК путем гомологичной рекомбинации. Распределение всех трех показателей (TMB, MSI, HRD) значительно отличалось от нормального, характеризовалось правосторонней асимметрией с выраженным эксцессом, что делает предпочтительным использование медианы и квартилей для оценки параметров распределения представленных величин в исследуемой популяции.
Мутационные сигнатуры были определены по всем мутациям, в том числе в некодирующей части генома. Несмотря на то, что отдельные мутации у пациентов сильно различаются, мутационные сигнатуры (паттерны нуклеотидных замен) у пациентов весьма схожи. Так, в исследуемой группе пациентов обнаружена активность 15 мутационных сигнатур (рис. 3). При этом значительный вклад в формирование мутаций (от 5 %) вносят только три из них – SBS1 (5 %), SBS5 (33 %) и SBS39 (57 %). Сигнатуры SBS1 и SBS5 являются clock-like сигнатурами, их накопление связано с возрастом. Этиология сигнатуры SBS39 не определена.
Обсуждение
В представленном исследовании проанализированы данные полногеномного секвенирования 71 образца колоректального рака и соответствующие им образцы клеток крови в качестве контроля. После фильтрации мутаций (SNP, Indel) было получе-
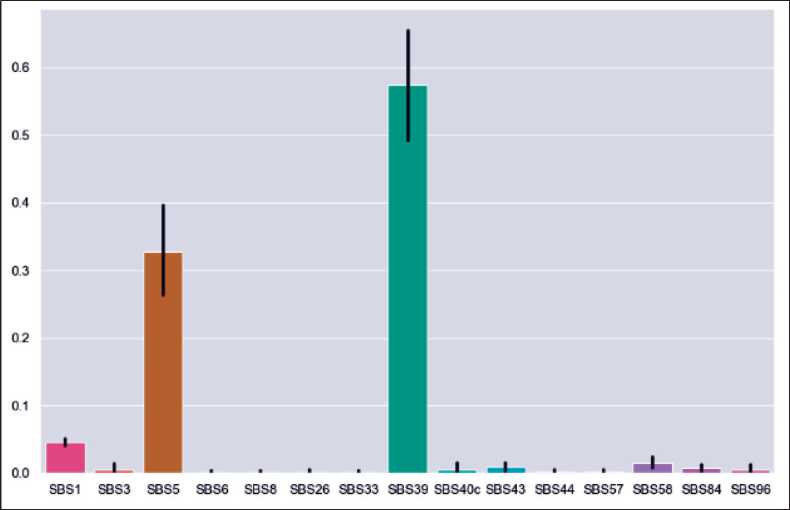
Рис. 3. Доля мутаций, относящихся к каждой мутационной сигнатуре с ненулевым вкладом. Горизонтальная ось – мутационные сигнатуры, вертикальная ось – средняя доля мутаций, которая относится к данной мутационной сигнатуре, по всем пациентам. Вертикальные линии обозначают 95 % ДИ; рисунок выполнен авторами
Fig. 3. Chart depicting the proportion of mutations belonging to each non-zero contribution mutation signature. The horizontal axis represents mutational signatures, the vertical axis represents the average proportion of mutations associated with each signature across all patients. Vertical lines indicate 95 % CI; created by the authors
но 22 275 соматических мутаций, 151 драйверная соматическая мутация, 70 маркерных соматических мутаций и 4 713 герминальных мутаций. Из них уникальными оказались 21 857, 111, 38, 405 мутаций соответственно. Выяснилось, что большая часть соматических мутаций являются уникальными, что свидетельствует о высокой мутационной гетерогенности исследуемой когорты.
В то же время мутации KRAS выявлены у 30 пациентов, что согласуется с литературными данными, указывающими на его роль как одного из наиболее часто мутирующих генов при развитии КРР [16]. Мутации BRAF V600E, хотя и менее частые, были обнаружены у четырех пациентов, что также свидетельствует о возможной важности этого маркера для прогноза заболевания и выбора терапии, особенно с учетом того, что BRAF V600E ассоциируется с более агрессивными формами некоторых злокачественных опухолей [17, 18].
При анализе CNV обнаружено, что в среднем примерно 20 % всех генов имеют измененное количество копий (амплификации и делеции примерно в равной пропорции). Среди CNV присутствуют амплификации важных онкогенов (например, BRAF ) и делеции онкосупрессоров (например, TP53 ). Тот факт, что амплификации и делеции происходят в равной пропорции, позволяет предположить, что такие изменения могут играть роль не только в активации онкогенов, но и в инактивации генов-супрессоров, что представляет собой важный аспект патогенеза КРР [19].
Мутационная нагрузка (TMB) в исследуемой когорте достаточно высокая (медианное значение TMB 5,5). Наблюдаемая высокая мутационная нагрузка может быть важным предсказателем клинического ответа на иммунотерапию, особенно с учетом того, что TMB выше 10 связано с улучшением прогноза при применении пембролизумаба [20, 21]. Однако, несмотря на высокую TMB, значения MSI и дефектов в системе репарации путем гомологичной рекомбинации были существенно низкими. Это может указывать на то, что в исследуемой когорте отсутствуют пациенты, которые могли бы получить пользу от терапии, направленной на дефекты репарации ДНК, такие как ингибиторы поли(АДФ-рибоза) полимеразы (PARP) [22, 23].
Профиль мутационных сигнатур во всех образцах очень схожий. Анализ мутационных сигнатур показал, что в исследуемой популяции преобладают мутационные сигнатуры SBS1, SBS5 и SBS39, что отражает возрастные изменения в клеточной ДНК, а также указывает на активность других, пока не уточненных, механизмов, связанных с сигнатурой SBS39 [24, 25]. Это открывает новые перспективы для дальнейших исследований в области молекулярной онкологии и подчеркивает важность детального анализа мутационных паттернов для выявления новых мишеней для терапии.
Заключение
Полученные результаты позволяют предположить, что мутационный процесс колоректального рака имеет многофакторный характер. Результаты проведенного исследования демонстрируют молекулярное разнообразие и генетическую гетерогенность опухолей у пациентов с колоректальным раком, а также подтверждают значимость ключевых генов и молекулярных изменений, таких как мутации в KRAS, BRAF, TP53, а также вариации числа копий генов в патогенезе заболевания. Вы- явленное многообразие изменений генома может быть связано с ошибками в репликации ДНК, воздействием внешних факторов и генетической предрасположенностью. Дальнейшие исследования помогут лучше понять эти механизмы и разработать новые подходы к диагностике, лечению и профилактике колоректального рака.
