Хроническая гранулематозная болезнь у ребенка двух лет: путь к диагнозу (клинический случай)
")
Автор: Спиваковский Ю.М., Щербина А.Ю., Спиваковская А.Ю., Юхачева Д.В., Першин Д.Е.
Журнал: Саратовский научно-медицинский журнал @ssmj
Рубрика: Педиатрия
Статья в выпуске: 3 т.18, 2022 года.
Бесплатный доступ
В статье представлен клинический случай диагностики хронической гранулематозной болезни у ребенка двух лет. Описаны сложности диагностики первичного иммунодефицитного состояния в рутинной педиатрической практике. Обсуждены проблемы диагностики первичных иммунодефицитов, определены неспецифические настораживающие признаки, когда педиатр должен в план дифференциальной диагностики включить первичный иммунодефицит.
Первичный иммунодефицит, хроническая гранулематозная болезнь
Короткий адрес: https://sciup.org/149141765
IDR: 149141765 | УДК: 616-002.7-036.12-07-053.4(045)
Chronic granulomatous disease in a two-year-old child: the trajectory to diagnosis (clinical case)
The article presents a clinical case of diagnosis of chronic granulomatous disease in a two-year-old child. The difficulties of diagnosing a primary immunodeficiency condition in routine pediatric practice are described. The problems of diagnosis of primary immunodeficiency are discussed, nonspecific warning signs are identified when a pediatrician should include primary immunodeficiency in the differential diagnosis plan.
Текст научной статьи Хроническая гранулематозная болезнь у ребенка двух лет: путь к диагнозу (клинический случай)
торов защиты принято называть термином «иммунодефицит» (ИД). Среди группы иммунодефицитов выделяют первичные (врожденные) (ПИД) и вторичные состояния [1]. По определению, ПиД — это многочисленная группа тяжелых заболеваний, имеющих генетическую обусловленность, причиной которых становится нарушение того или иного звена иммунитета. Научная группа Всемирной организации здра- воохранения к настоящему моменту выделила более 350 нозологических форм ПИД, Международным союзом иммунологических сообществ (International Union of Immunological Societies) признана связь первичных иммунодефицитов почти с четырьмя сотнями различных генов, при этом идентифицировано несколько тысяч мутаций в них, и процесс их поиска и фиксации продолжается [2, 3]. Данные Международного фонда первичных иммунодефицитов (Jeffry Modell Foundation) свидетельствуют о том, что на планете как минимум 10 млн человек страдают той или иной формой ПИД [4].
Классификация 2019 г., предложенная Международным союзом иммунологических обществ (International Union of Immunologic Societies — IUIS), делит ПИД на 10 основных групп, многие из которых, в свою очередь, подразделяются на подгруппы. Дополнение общего регистра заболеваний, относящихся к группе первичных иммунодефицитов, происходит практически ежегодно [3, 5].
Несмотря на столь бурное развитие иммунологии, и в частности раздела по изучению первичных иммунодефицитных состояний, их диагностика в рутинной клинической практике остается крайне сложной для врача задачей. Очень важным в этом вопросе является умение заподозрить ИД по совокупности клинических симптомов, течению интеркуррентных заболеваний и иным клиническим маркерам. С этой целью специалистами сообщества иммунологов были предложены ориентировочные («настораживающие») признаки, когда педиатр должен включить первичные иммунодефицитные состояния в группу дифференциально-диагностического поиска [6]. Однако, несмотря на столь серьезные исследования в данной области, своевременная диагностика ПИД испытывает значительные сложности как в России, так и в других странах, что часто связано с редкостью встречаемости патологии и недостаточной настороженностью врачей первичного контакта [2, 7].
В актуальных обзорах, основанных на отечественном регистре больных с первичными иммунодефицитными состояниями, указывается, что распространенность данных состояний в Российской Федерации в 2019 г. составляла 1,35 человека на 100 тыс. населения, что значительно уступает европейским данным [7].
Представляемый клинический случай показывает непростой путь диагностики одного из видов первичного иммунодефицитного состояния у ребенка двух лет. Все данные о течении заболевания публикуются с разрешения законных представителей ребенка.
Описание клинического случая. Мальчик П., 1 г. 11 мес., осмотрен на консультативном приеме в Клинической больнице им. С. Р. Миротворцева СГМУ в середине мая 2020 г. в связи с жалобами родителей на выраженный отек правого глаза, преимущественно в области верхнего века.
При уточнении анамнеза выяснено, что ребенок от беременности, протекавшей с угрозой прерывания в первом триместре, многоводием — в третьем триместре. Роды происходили в срок, без патологии. В возрасте 3 мес. проводилось зондирование слезных каналов с обеих сторон в связи с длительно текущим дакриоциститом новорожденного, в 7 мес. осуществлялось повторное зондирование с одной стороны. Данные о посеве на флору отделяемого из слезного мешка отсутствуют. В июле 2019 г. (1 г. 1 мес.) была госпитализация в детский инфекционный стационар по месту жительства в связи с длительной лихорад- кой, не сопровождавшейся катаральными проявлениями. В этот период выявлена анемия (гемоглобин 79 г/л). Выставлен диагноз острого респираторного заболевания, проведен курс пероральной антибактериальной терапии азитромицином с хорошим клиническим эффектом. В последующем ребенок продолжал получать препараты железа с оценкой эффекта по показателям гемоглобина, с достижением через месяц наблюдения уровня 105 г/л. В возрасте одного года отмечен гиперэргический результат пробы Манту (18 мм), по поводу чего мальчик консультирован и обследован фтизиатром.
Впервые выраженную припухлость верхнего века правого глаза ребенка родители отметили 13.02.2020. После амбулаторной консультации офтальмологом пациент получал местные топические глюкокортикостероиды, антигистаминные препараты в связи с подозрением на аллергический генез отека. По результатам ультразвукового исследования глазницы патологических новообразований выявлено не было. На фоне местной терапии отмечено ухудшение состояния в виде нарастания отека, гиперемии глазного яблока и века. Повторно консультирован и наблюдался офтальмологом и оториноларингологом. В ходе дообследования были исключены травматическое повреждение глаза, его инфекционное поражение, воспалительные изменения ЛОР-органов. В связи с сохранением жалоб, которые носили переменный характер даже в течение дня (нарастание и уменьшение отека), ребенок в марте 2020 г. был госпитализирован первоначально в неврологическое, а в последующем в педиатрическое отделение областной детской больницы с диагнозом «Рецидивирующий ангионевротический отек век справа». В ходе обследования и наблюдения в стационаре по результатам лабораторных тестов наблюдалось: в общем анализе крови — сохранение анемии (гемоглобин 89 г/л), лейкоцитов — 10,2х109/л, лейкоцитарная формула: эозинофилы 2%, базофилы 1 %, палочкоядерные нейтрофилы 5%, сегментоядерные нейтрофилы 37%, лимфоциты 50%, моноциты 5%, скорость оседания эритроцитов 13 мм/час; в биохимическом анализе крови — без значительных отклонений от нормы на фоне повышенного уровня С -реактивного белка до 24 мг/л (норма — менее 6 мг/л); гипергаммаглобулинемия — уровень иммуноглобулинов класса G 2268 мг/дл (норма до 1200 мг/дл). Проведена магнитно-резонансная томография (МРТ) головного мозга, где описана картина единичного очагового изменения в веществе головного мозга, вероятно, резидуального характера, МР-картина структурных изменений круговой мышцы глаза, височной мышцы справа, по всей вероятности, воспалительного характера. В ходе комплексного обследования проводилась компьютерная томография грудной клетки, где выявлен ателектаз S4 правого легкого, на фоне его уплотнения и описаны деформированные бронхи.
В связи с выявленными изменениями проведена диагностическая бронхоскопия, по результатам которой выявлены справа в области устья бронха средней доли рубцовые изменения слизистой в виде истончения шпоры устья. При санации получено небольшое количество вязкой слизистой мокроты, которая взята для бактериологического исследования (получен отрицательный результат). Исходя из отсутствия значимой респираторной симптоматики проведено углубленное фтизиатрическое обследование, не выявившее значимой патологии. Ребенок, кроме того, был осмотрен иммунологом, оториноларингологом, гематологом.
С учетом полученных данных о бронхолегочных изменениях в дополнение к противоаллергической терапии был проведен курс азитромицином в течение пяти дней. По основной жалобе описано некоторое улучшение, выражающееся в уменьшении отека правого века, на фоне чего ребенок выписан из стационара с диагнозом: «Рецидивирующий ангионевротический отек век справа, персиcтирующее течение». Сопутствующие заболевания: «Атопический дерматит, младенческая форма, персиcтирующее течение. Аллергия к белкам коровьего молока (клинически). Железодефицитная анемия средней степени тяжести смешанного генеза. Ателектаз S4 правого легкого».
После выписки из стационара состояние ребенка оставалось стабильным, сохранялся умеренный отек правого века, проявления которого продолжали носить волнообразный характер. Во второй половине мая 2020 г. на фоне сохраняющегося с тенденцией к усилению отека века правого глаза мальчик был консультирован в Клинической больнице имени С. Р. Миротворцева СГМУ; в контрольных анализах крови отмечена отрицательная динамика, что и стало поводом к госпитализации в педиатрическое отделение.
При поступлении, 20.05.2020, состояние расценено как среднетяжелое. Температура тела при трехчасовом мониторинге — в пределах нормальных значений. Вес 15,8 кг, рост 89 см, ИМТ 20,2.
Объективно при осмотре: ребенок в сознании, контактен, на осмотр реагирует адекватно. Кожные покровы бледные, отмечается сухость и гиперемия кожи щек, на коже бедер — единичные элементы в виде пятен гиперемии, местами со следами расчесов. Отечность и умеренная гиперемия век правого глаза. Носовое дыхание свободное. Зев без особенностей. Периферические лимфоузлы не увеличены. Костно-мышечная система без особенностей, движения в суставах в полном объеме. Прекардиаль-ная область не изменена. Границы относительной сердечной тупости соответствовали возрастной норме. Тоны сердца ясные, ритмичные. Грудная клетка нормостеническая, обе ее половины симметрично участвуют в акте дыхания; при перкуссии над легочными полями — ясный легочный звук. Аускультатив-но дыхание пуэрильное, хрипы не выслушиваются. Живот обычной формы, доступен глубокой пальпации, безболезненный. Печень не увеличена. Селезенка не пальпируется. Мочевыделительная система при физикальном обследовании без отклонений от нормы.
Данные лабораторного обследования: в общем анализе крови (при повторных исследованиях): эритроциты — в пределах 4,98-4,49х1012/л; лейкоциты — от 18,9 до 9,6х109/л; гемоглобин 91 г/л; тромбоциты — от 494 до 605х109/л; скорость оседания эритроцитов — от 29 до 32 мм/час; во всех анализах профиль лейкоцитарной формулы сохранялся лимфоцитарным.
В биохимическом анализе крови: все основные показатели в пределах референтных значений; в динамике на фоне антибактериальной терапии отмечено нарастание уровня трансаминаз (аланинаминотрансферазы — до 180 Е/л и аспартатаминотрансферазы — до 80 Е/л), а также щелочной фосфатазы — до 1173 Е/л. Уровень С -реактивного белка был повышен до 10-14 мг/л (норма до 5 мг/л).
По результатам ультразвукового исследования мягких тканей правого глаза: в верхненаружном углу орбиты выявлено гипоэхогенное образование с сосудистой сетью размерами 9,26×24,21 мм.
По результатам компьютерной томографии органов грудной полости была описана картина правосторонней полисегментарной пневмонии с признаками гиповентиляции S5 средней доли правого легкого и правосторонним экссудативным плевритом.
При ретроспективной оценке ранее проведенного МРТ дополнительно к уже описанным изменениям выявлены признаки резорбции скулового отростка лобной кости, в соответствии с чем была проведена компьютерная томография орбит и костей черепа, где описана картина новообразования правой слезной железы с изменениями периорбитальной клетчатки и резорбцией правого скулового отростка лобной кости.
С учетом выявленных изменений комплекс обследований был дополнен исследованиями, направленными на возможное установление генеза опухолевидного образования глазницы. Были проведены серологические исследования для выявления маркеров глистнопаразитарного поражения, онкомаркеров, остеосцинтиграфия, исследование антител к цитоплазме нейтрофилов (выявлены положительные антитела к BPI (бактерицидному белку), эластазе, катепсину G), комплексное бактериологическое обследование, консультация онколога.
При оценке результатов комплексного обследования в сочетании с оценкой эффекта от проводимой антибактериальной терапии было отмечено, что обнаруженные изменения в легких не коррелировали с локальной клинической симптоматикой, что позволяло расценивать их на тот момент как возможное проявление неопластического процесса. Выявленные в биохимическом анализе крови изменения в виде повышения уровня трансаминаз и щелочной фосфатазы, видимо, могли быть обусловлены цитотоксическим эффектом новообразования, сопровождающимся деструкцией костной ткани.
При повторных консультативных осмотрах в специализированной университетской офтальмологической клинике в опухолевидном образовании слезной железы было заподозрено течение аденокистозного рака.
Таким образом, по результатам наблюдения педиатра, онколога, офтальмолога было высказано мнение о необходимости скорейшей, в том числе морфологической, верификации новообразования правой орбиты, с учетом подозрения на злокачественную природу опухоли. В связи с этим было рекомендовано дообследование в условиях федерального центра по профилю «онкология».
Диагноз при выписке (29.05.2020) и направлении в ФГБУ «Национальный медицинский исследовательский центр онкологии им. Н. Н. Блохина» Минздрава России (НМИЦ онкологии им. Н. Н. Блохина) определен следующим образом: «Основной: Новообразование правой орбиты (С69.6). Сопутствующие заболевания: Внебольничная полисегментарная пневмония с локализацией в S5, S9–10 справа, осложненная ателектазом S5, правосторонним экссудативным плевритом (не исключается, как следствие основного заболевания). Железодефицитная анемия, средней степени тяжести, смешанного генеза. Атопический дерматит, младенческая форма, период неполной ремиссии. Избыток массы тела. Малые аномалии развития сердца».
Дальнейшее обследование на базе научноконсультативного отделения НМИЦ онкологии им. Н. Н. Блохина было осложнено тем, что на фоне сохраняющихся односторонних изменений в правом легком, которые описывались как пневмония, обследование с использованием анестезиологического пособия оказалось невозможным. Вместе с тем в амбулаторных условиях и при продолжении антибактериальной терапии (защищенные цефалоспорины IV поколения, аминогликозиды, макролиды) проведены контроль МРТ головного мозга, МРТ области правой глазницы с контрастированием, повторно — компьютерная томография грудной клетки и дополнительное ультразвуковое обследование. По данным МРТ орбиты — в правой височной области, верхнелатеральных отделах правой орбиты наблюдались извитые мелкие округлые жидкостные образования размером 20×14×18 мм, накапливающие контраст; выявлен дефект лобной кости 25×8 мм без признаков склероза в области верхнелатерального края правой орбиты. Отрицательной динамики размеров образования по сравнению с предыдущим исследованиями не было выявлено. По результатам состоявшегося этапного консилиума был сделан предварительный вывод: с учетом того, что процесс носит специфически очаговый характер, у ребенка с высокой вероятностью имеется гранулематоз Вегенера.
По данному поводу 17.06.2020 пациент был осмотрен и проконсультирован врачом-ревматологом ФГАУ «Национальный медицинский исследовательский центр здоровья детей» Минздрава России, где для верификации диагноза было рекомендовано проведение биопсии патологического образования правой глазницы и последующее морфологическое исследование.
Проведена госпитализация в НИИ Детской онкологии и гематологии НМИЦ онкологии им. Н. Н. Блохина, где проведено дополнительное клинико-лабораторное обследование, в том числе верхняя транскутанная орбитотомия справа с биопсией мягких тканей и надкостницы верхнелатерального отдела орбиты. При гистологическом исследовании ни в одном из взятых при биопсии фрагментов опухолевый рост выявлен не был. Наряду с этим по результатам иммунологического исследования крови были выявлены изменения в виде снижения цитотоксического потенциала CD16+- и CD8+-лимфоцитов. По результатам консилиума, принимая во внимание длительно текущие пневмонии в анамнезе, рецидивирующий отек верхнего века, иммунологические изменения, а также умеренную положительную динамику (по данным инструментального контроля) на фоне проводимой неспецифической антибактериальной терапии, сделано заключение о том, что патологическое состояние ребенка следует дифференцировать между проявлениями системного иммунопатологического процесса и дебютом эозинофильного гранулематоза.
Последующее обследование проведено на базе ФГБУ «Национальный медицинский исследовательский центр детской гематологии, онкологии и иммунологии им. Дмитрия Рогачева» Минздрава России (НМИЦ ДГОИ им. Дмитрия Рогачева). В условиях специализированного стационара в результате комплексного обследования, и в частности по результатам проведенного BURST-теста (лабораторного анализа по оценке цитотоксической [бактерицидной] активности нейтрофилов и моноцитов, которая опос-
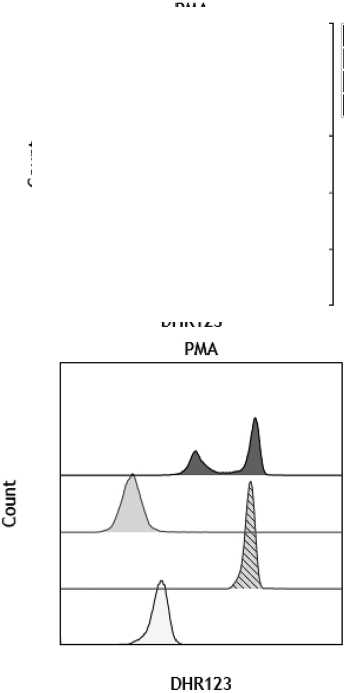
SI здоровый донор 144,7 (>30)
SI low мать пациента 11,7 (<30)
SI high мать пациента 233,5 (>30)
Бимодальный характер флюоресценции у матери пациента
□стимуляция мать пациента
□негативный контроль мать пациента
□стимуляция здоровый донор
□негативный контроль здоровый донор
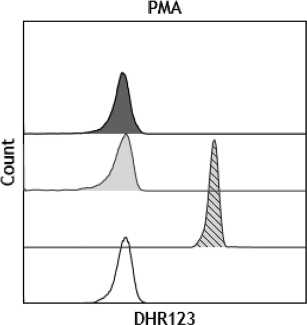
■стимуляция пациент
□негативный контроль пациент
□стимуляция здоровый донор
□ негативный контроль здоровый донор
Результаты BURST-теста пациента П. и его матери.
Основы трактовки результатов исследования (представлено из базы данных лаборатории трансплантационной иммунологии и иммунотерапии гемобластозов
ФГБУ «НМИЦ ДГОИ им. Дмитрия Рогачева»)
редована протеолитическими ферментами их гранул и активными формами кислорода) (рисунок), было выявлено снижение окислительной способности нейтрофилов, был установлен диагноз «Хроническая гранулематозная болезнь» (ХГБ).
В основе диагностики лежит оценка окислительного взрыва нейтрофилов после стимуляции в образе гепаринизированной крови. В результате окисления дигидрородамина (DHR 123) определяется интенсивность флюоресценции родамина 123 с помощью проточной цитометрии.
Индекс стимуляции (SI) является отношением интенсивности флюоресценции DHR 123 в стимулированных и нестимулированных нейтрофилах.
Окислительный взрыв расценивается как нормальный при значении SI>30. При наличии Х-сцепленной мутации у пациентов женского пола наблюдается бимодальный характер флюоресценции.
При обследовании матери мальчика выявлено бимодальное распределение окислительной способности нейтрофилов, что позволяет с высокой вероятностью предположить Х-сцепленную форму заболевания. С целью выявления специфических мутаций, характерных для данного заболевания, кровь ребенка была отправлена для проведения прямого секвенирования по Сэнгеру. В стационаре начата терапия вориконазолом и Ко-тримоксазолом, которую рекомендовано проводить постоянно с контролем эффективной дозы препаратов.
В последующем, по результатам проведенного в лаборатории молекулярной биологии НМИЦ ДГОИ им. Дмитрия Рогачева исследования было установлено, что в экзоне 7 гена CYBB выявлена гемизи-готная замена c.676C>T, приводящая к возникновению преждевременного терминирующего кодона p.(Arg266Ter). Данный вариант неоднократно описан в литературе как патогенный при ХГБ [4].
При последующих повторных госпитализациях в НМИЦ ДГОИ им. Дмитрия Рогачева на фоне проводимой терапии и при постоянных лабораторном и инструментальном наблюдениях было принято решение о проведении трансплантации гемопоэтических стволовых клеток. Данная высокотехнологичная процедура была проведена примерно через 9 мес. с момента первичной клинической верификации диагноза. Однако в последующем появились показания к повторному проведению трансплантации гемопоэтических стволовых клеток в связи с развившимися явлениями гипофункции трансплантата. Ребенок продолжает наблюдаться и получать лечение в НМИЦ ДГОИ им. Дмитрия Рогачева.
Обсуждение. ХГБ характеризуется нарушением функциональной активности фагоцитов (образованием активных форм кислородных радикалов, внутриклеточного киллинга и фрагментации фагоцитированных патогенов), постоянными бактериальными и грибковыми инфекционными заболеваниями и возникновением гранулематозного воспаления [8]. Развитие этой болезни может быть обусловлено дефектами любой из пяти субъединиц комплекса НАДФН-оксидазы, ответственных за респираторный взрыв фагоцитарных лейкоцитов.
Первое упоминание ХГБ у детей относится 1959 г., когда эта патология была описана как фатальная [9]. Болезнь не поддавалась лечению; на аутопсии выявляли генерализованное гранулематозное воспаление, отличающееся по своей специфике от известных на тот момент инфекционных и неинфекционных заболеваний. К настоящему моменту ХГБ достаточно хорошо изучена, и процесс накопления данных непрерывно продолжается, в том числе выявляются новые варианты генетических поломок, приводящих к формированию типичной клинической картины заболевания [3, 10].
ХГБ имеет наследственную природу. Частота встречаемости этой патологии варьирует, по данным разных авторов, от 1 на 250000 до 1 на 4000000 в США и 8,5 на 1000000 в Великобритании [11], это позволяет отнести ХГБ к достаточно редким заболеваниям, что, в свою очередь, создает дополнительные трудности при ее диагностике. Не менее ⅔ всех случаев ХГБ относятся к вариантам X-сцепленного наследования, а остальные — к аутосомно-рецессивным [12].
Отличительной чертой клинических проявлений ХГБ являются рецидивирующие инфекционные заболевания. В силу особенностей контакта организма человека с факторами внешней среды основными локусами инфекционных поражений следует считать инфекции кожи, легких и кишечника. В связи с отсутствием особенностей клинического течения подобных инфекционных заболеваний единственной опорой врача в формировании подозрения на наличие у пациента ХГБ могут стать только прогрессирование или резистентность течения заболевания на фоне стандартной терапии [13].
В представленном клиническом случае трудность постановки диагноза была связана с различными причинами. Например, достаточно сложно было предположить, что проявления ангионевротического отека были обусловлены не аллергической или иной подобной причиной, а вероятным лимфостазом на фоне сформировавшейся гранулемы. Между тем односторонность процесса, цвет кожи в области зоны первичной симптоматики, ее температура, длительность проявлений позволяли усомниться в аллергическом генезе заболевания.
Серьезным поворотным пунктом диагностического поиска в обсуждаемом случае стало обнаружение очага костной деструкции в области скулового отрост- ка лобной кости. Преобладающей диагностической доктриной в этот момент стал поиск неопластического или специфического (туберкулезного) процесса. В то же время в отечественной и зарубежной литературе описанию фактов обнаружения деструктивных костных поражений при ПИД посвящен ряд работ, где при ХГБ в качестве приоритетного симптома указываются местная реакция и лимфаденит в ответ на противотуберкулезную вакцинацию [14]. В представленном нами клиническом случае костная деструкция имела иной генез, а признаков микобактериальной инфекции при повторных обследованиях выявлено не было.
Отмеченные у маленького пациента легочные поражения также сфокусировали внимание на поиске стандартных неспецифических воспалительных процессов в легочной ткани, осложненных явлениями гиповентиляции в области одного сегмента. У больных старшего возраста легочные поражения — это чаще всего ведущее клиническое проявление ХГБ, а у пациентов младшего возраста — специфическое грибковое поражение легочной ткани [15].
Несмотря на бурное развитие генетики, включая появление возможности проведения углубленных генетических исследований в максимально ранние сроки, сохраняются наиболее типичные возрастные интервалы, когда данный диагноз может быть заподозрен в связи с началом клинически значимой манифестации. Нашему пациенту, даже при нетипичности симптоматики, к моменту постановки диагноза было немногим более двух лет, что, по данным литературы, и есть наиболее частый возраст постановки подобного диагноза [8]. Вместе с тем следует отметить, что ПИД при проведении диагностического поиска не фигурировал среди списка наиболее вероятных диагнозов.
Обсуждая проблему пути к диагнозу в рамках педиатрической аудитории считаем уместным еще раз обратиться к общеизвестным признакам, когда диагноз ПИД может быть заподозрен на основании данных анамнеза [5]: а) более четырех отитов в год; б) более двух синуситов в год; в) проведение более 2 мес. антибактериальной терапии с минимальным эффектом, необходимость назначения внутривенных антибиотиков для разрешения инфекции; г) более двух пневмоний в год; д) повторные абсцессы кожи и внутренних органов; е) упорная молочница у лиц старше одного года; ж) более двух тяжелых инфекционных процессов (сепсис, остеомиелит, менингит и др.); з) оппортунистические инфекции (вызываемые Pneumocystis jirovecii и другими возбудителями); и) отставание младенца в весе на фоне повторных эпизодов диареи; к) наличие в семейном анамнезе смертей в раннем возрасте, с клиникой инфекционных заболеваний или выявленного иммунодефицитного состояния.
Нельзя сказать, что наш пациент в полной мере соответствовал какому-либо из представленных критериев, однако при определенной настороженности некоторые из фактов его клинического анамнеза могли бы навести на мысль о необходимости проведения дополнительного диагностического поиска в этом направлении.
Сегодня в медицинской литературе имеется очень большое количество информации, позволяющей рассматривать проблемы ХГБ в самых разных аспектах. Опубликованные различные варианты систематических обзоров и описанные отдельные клинические случаи позволяют в должной мере представить частоту различных мутаций, их связь с этническими группами, варианты клинических манифестных признаков, эффективность различных видов терапии, а также обсуждать прогноз заболевания [16], часть работ посвящена случаям сочетания ХГБ с аутоиммунными заболеваниями [17], сложной гематологической патологией [18], а также у пациентов с «нетипичным» возрастом манифестации [19].
На современном этапе развития медицины в качестве наиболее эффективного метода лечения указывается на необходимость применения трансплантации гемопоэтических стволовых клеток при ХГБ. И если в обзорах, относящихся последнему десятилетию XX столетия и к началу XXI в. еще указывалось на ограниченное число пациентов, получающих терапию по данной методике, то в настоящее время отечественные специалисты представляют сопоставимое число проведенных подобных высокотехнологичных вмешательств только на базе одного крупного национального центра [20]. Авторы подобных обзоров указывают на значительно возросший процент эффективности применения трансплантации гемопоэтических стволовых клеток у пациентов с ХГБ, что связано с разработкой новых таргетных препаратов, совершенствованием технологических приемов, улучшением методик кондиционирования, и делают вывод о том, что данная методика в перспективе будет совершенствоваться и результаты ее применения будут неизменно улучшаться. Это, в свою очередь, будет увеличивать продолжительность и качество жизни пациентов с ХГБ.
Таким образом, представленный клинический случай наглядно продемонстрировал имеющиеся проблемы, возникающие на этапах диагностики ПИД в рутинной педиатрической практике. Особенность случая требует дополнительной настороженности со стороны врача-педиатра при обследовании пациента, имеющего любые из ориентировочных признаков, при которых должен быть заподозрен ПИД или иное нетипично текущее заболевание, особенно в младшем детском возрасте.
Список литературы Хроническая гранулематозная болезнь у ребенка двух лет: путь к диагнозу (клинический случай)
- Delyagin VM, Sadovnikova IV. Primary immunodeficiency in pediatric practice. Moscow: GEOTAR-Media, 2020; 80 p. Russian (Делягин В. М., Садовникова И. В. Первичные иммунодефициты в педиатрической практике. М.: ГЭОТАР-Медиа, 2020; 80 с.).
- Rezaei N, Pandey S, Harville T, et al. Introduction to Primary Immunodeficiencies. In: Rezaei N., ed. Pediatric Immunology. Springer Nature Switzerland AG, 2019; 822 p. DOI: 10.1007 / 978‑3‑030‑21262‑9_1.
- Bousfiha A, Jeddane L, Picard C, et al. Human Inborn Errors of Immunity: 2019 Update of the IUIS Phenotypical Classification. J Clin Immunol 2020; 40 (1): 66–81. DOI: 10.1007 / s10875‑020‑00758‑x.
- Modell V, Gee B, Lewis DB, et al. Global study of primary immunodeficiency diseases (PI) — diagnosis, treatment, and economic impact: an updated report from the Jeffrey Modell Foundation. Immunol Res 2011; 51 (1): 61–70.
- Shcherbina AYu. Masks of primary immunodeficiency disorders: diagnostic and therapeutic problems. Russian Journal of Pediatric Hematology and Oncology 2016; 3 (1): 52–8. Russian (Щербина А. Ю. Маски первичных иммунодефицитных состояний: проблемы диагностики и терапии. Российский журнал детской гематологии и онкологии 2016; 3 (1): 52–8). DOI: 10.17650 / 2311‑1267‑2016‑3‑1‑52‑58.
- Conley ME, Notarangelo LD, Etzioni A. Diagnostic criteria for primary immunodeficiencies. Representing PAGID (Pan-American Group for Immunodeficiency) and ESID (European Society for Immunodeficiencies). Clin Immunol 1999; 93 (3): 190–7. DOI: 10.1006 / clim.1999.4799.
- Mukhina АА, Kuzmenko NB, Rodina YA, et al. Characteristics of patients with primary immunodeficiency states in the Russian Federation: from birth to old age. Pediatria n. a. G. N. Speransky 2019; 98 (3): 24–31. Russian (Мухина А. А., Кузьменко Н. Б., Родина Ю. А. и др. Характеристика пациентов с первичными иммунодефицитными состояниями в Российской Федерации: от рождения до старости. Педиатрия им. Г. Н. Сперанского 2019; 98 (3): 24–31).
- Khaitov RM. Immunology: textbook. 2nd ed., corr. and suppl. Moscow: GEOTAR-Media, 2013; 528 p. Russian (Хаитов Р. М. Иммунология: учебник. 2‑е изд., испр. и доп. М.: ГЭОТАР-Медиа, 2013; 528 с.).
- Bridges RA, Berendes H, Good RA. A fatal granulomatous disease of childhood; the clinical, pathological, and laboratory features of a new syndrome. AMA J Dis Child 1959; 97 (4): 387–408.
- Stray-Pedersen A, Sorte HS, Samarakoon P, et al. Primary immunodeficiency diseases: genomic approaches delineate heterogeneous Mendelian disorders. J Allergy Clin Immunol 2017; 139 (1): 232–45. DOI: 10.1016 / j.jaci.2016.05.042.
- Klimenko VA, Piontkovska OV, Lupaltseva OS, et al. Chronic Crohn’s disease: diagnosis and management at the present times. Zdorov’e Rebenka 2018; 13 (1): 115–22. Russian ( [Клименко В. А., Пионтковская О. В., Лупальцова О. С. и др. Хроническая гранулематозная болезнь: диагностика и ведение больных на современном этапе]. Zdorov’e Rebenka 2018; 13 (1): 115–22 [на укр. яз.]). DOI: 10.22141 / 2224-0551.13.1.2018.127074.
- Chiriaco М, Salfa I, Di Matteo G, et al. Chronic granulomatous disease: Clinical, molecular, and therapeutic aspects. Review Pediatr Allergy Immunol 2016; 27 (3): 242–53. DOI: 10.1111 / pai.12527.
- Chernysheva I, Volokha A, Bondarenko AB, et al. Chronic granulomatous disease: the experience of diagnosis and treatment in children. Child’s Health 2013; 4 (47): 61–6. Russian ([Чернышева И., Волоха А., Бондаренко А. В. и др. Хроническая гранулематозная болезнь: опыт диагностики и лечения у детей. Здоровье ребенка] 2013; 4 (47): 61–6 [на укр. яз.]).
- Conti F, Lugo-Reyes SO, Blancas Galicia L, et al. Mycobacterial disease in patients with chronic granulomatous disease: a retrospective analysis of 71 cases. Journal of Allergy and Clinical Immunology 2016; 138 (1): 241–8. DOI: 10.1016 / j.jaci.2015.11.041.
- Bondarenko AV, Chernishova LI, Kostyuchenko LV, et al. Mycoses in the structure of infectious syndrome in primary immunodeficiency. Sovremennaya Pediatriya 2015; 4 (68): 96. Russian (Бондаренко А. В., Чернишова Л. И., Костюченко Л. В. и др. Микозы в структуре инфекционного синдрома при первичных иммунодефицитах. Современная педиатрия 2015; 4 (68): 96). DOI: 10.15574 / SP. 2015.68.96.
- Serebryakova EN, Volosnikov DK, Pischalnikov AYu, et al. Chronic granulomatous disease: A literature review and a description of cases of chronic granulomatous disease in children of the Chelyabinsk Region. Trudnyj Pacient = Difficult Patient 2016;
- 14 (2-3): 46–50. Russian (Серебрякова Е. Н., Волосников Д. К., Пищальников А. Ю. и др. Хроническая гранулематозная болезнь: обзор литературы и описание случаев хронической гранулематозной болезни у детей Челябинской области. Трудный пациент 2016; 14 (2-3): 46–50).
- Gargouri L, Safi F, Mejdoub I, et al. Auto-immune hepatitis in chronic granulomatous disease in a 2‑year-old girl. Archives de Pediatrie 2015; 22 (5): 518–22. DOI: 10.1016 / j.arcped. 2015.02.003.
- Valentine G, Thomas TA, Nguyen T, Lai YC. Chronic granulomatous disease presenting as hemophagocytic lymphohistiocytosis: A case report. Pediatrics 2014; 134 (6): e1727–30. DOI: 10.1542 / peds.2014-2175.
- Miladinovic M, Wittekindt B, Fischer S, et al. Case report: symptomatic chronic granulomatous disease in the newborn. Front Immunol 2021; (12): 663883. DOI: 10.3389 / fimmu. 2021.663883.
- Machneva EB, Pristanskova EA, Olkhova LV, et al. Results of allogeneic hematopoietic stem cell transplantation in patients with chronic granulomatous disease in the Russia children’s clinical hospital nice. Russian Journal of Pediatric Hematology and Oncology 2020; 7 (2): 23–34. Russian (Мачнева Е. Б., Пристанскова Е. А., Ольхова Л. В. и др. Результаты аллогенной трансплантации гемопоэтических стволовых клеток у пациентов с хронической гранулематозной болезнью в Российской детской клинической больнице. Российский журнал детской гематологии и онкологии 2020; 7 (2): 23–34. DOI: 10.21682 / 2311‑1267‑2020‑7‑2‑23‑34.
