In-silico docking studies of calendoflavobioside of Calendula officinalis Linn. against matrix metalloproteinase 8 and 12 on wound healing

Author: Gunasekaran Shobana, Nayagam Agnel Arul John
Journal: Журнал стресс-физиологии и биохимии @jspb
Article in issue: 3 т.21, 2025.
Free access
Bioactive compounds are a rich source of preventive substances for many diseases. In the present study was attempt to evaluate the in silico docking studies of calendoflavobioside of Calendula officinalis Linn. on wound healing. The compound calendoflavobioside were screened for the wound healing biomarker matrix metallo proteinases 8 and 12 by molecular docking. The result of the study indicates that calendoflavobioside has good affinity with MMP 8(-3.00kcal/mol) and MMP 12(-3.08kcal/mol).
Metallo proteinase 8, metallo proteinase 12, calendula officinalis, calendoflavobioside, molecular docking
Short address: https://sciup.org/143184727
IDR: 143184727
Text of the scientific article In-silico docking studies of calendoflavobioside of Calendula officinalis Linn. against matrix metalloproteinase 8 and 12 on wound healing
Wound can be defined as a break in the cellular and anatomical architecture of body tissue that includes skin, mucus membrane, deeply lying tissues or surface of internal organs ranging from incision, laceration, abrasion, puncture, and closed wounds such as contusion, hematoma and crush (Adiele et al ., 2014). Wound healing is a restorative response by the tissue during injury. It involves the complex cascade of cellular events that includes three faces, namely resurfacing, reconstitution, and restoration of the tensile strength of injured skin. Often healing process is explained in four terms with each classic phase overlapping each other: Hemostasis, inflammation, proliferation and maturation.
Natural products represent a rich source of biologically active compounds and are an example of molecular diversity, with recognized potential in drug discovery and development. Despite changing strategies in natural product research, which included sample selection and collection, isolation techniques, structure elucidation of the isolates, biological evaluation, biosynthesis have been increased, so the rate of discovery of truly novel natural product drugs were also increased. The advent of bioinformatics reduced the cost and time of drug screening process. In an effort to reduce the cost of developing new medicines and their time to market, pharmaceutical companies have attempted to streamline the drug discovery process using computational methods. Today, virtually every drug company of appreciable size has adopted computational methodology in most stages of the design process (Jorgensen 2004; Barril and Soliva 2006; Tramontano 2006) . Many computational methods complement one another and may be combined to help rationalize the drug discovery process. In cases where it is possible to determine the 3-dimensional structure of the biomolecular target, molecular docking (Brooijmans and Kuntz 2003; Sousa et al . 2006) becomes possible and allows structure based hit identification and/or lead optimization (Shoichet et al . 2002; Kitchen et al . 2004) .
Molecular docking is an in-silico computational technique used to predict conformation and binding affinity of intermolecular complexes based on the three- dimensional structures of individual molecules. This method is widely used in the field of structure-based drug design, in which researchers try to find compounds, which will form a stable intermolecular complex with a target protein. The target protein is usually known to play a vital role in a pathological process, so finding a potent inhibitor is crucial in disruption of its function. Initial screening of possibly millions of compounds in a laboratory conditions is often too expensive and timeconsuming process to be feasible and thus fast molecular docking methods are used to eliminate unlikely candidates (Kitchen et al., 2004). ike many methods in the field of computational chemistry, molecular docking uses a number of approximations to reduce a time required for each simulation. As it has many applications, it is important to aware of its accuracy and possible limitations.
One of the identified challenges is how docking software handles non-standard atoms, such as metal atoms (Irwin et al ., 2005) . While docking programs often employ chemical system descriptions based on molecular mechanics, majority of them requires parameters for individual atom types. An extensive effort has been put into optimization of these parameters for common atom types, such as atom types of carbon and oxygen. However, parameters for non-standard atoms are usually based on a much smaller data sets, or entirely missing in the software.
Matrix metalloproteinase (MMP) is a family of zincdependent, calcium-containing endopeptidases. The MMPs belong to a larger family of proteases known as metzincin super family (Gomis-Ruth, 2003). MMPs can be divided into eight groups based on their domain structure (Egeblad and Werb, 2002). Three common homologous domains include the N-terminalpro-peptide, the catalytic domain and the hemopexin-like C-terminal domain, which is linked to the catalytic domain by a flexible hinge region.
MMPs perform a variety of roles in living organisms. They are responsible for the tissue remodeling and degradation of many extracellular matrix (ECM) proteins, including collagens, elastins, gelatin, matrix glycoproteins and proteoglycan. More recently, it has also been recognized that they cleave many other peptides and proteins and perform other functions that may be independent of proteolytic activity. Physiological processes where MMPs are involved include angiogenesis (formation of new blood vessels), apoptosis (process of programmed cell death), bone modeling or wound healing (Overall et al., 2002).
Under normal physiological conditions, level of MMP expression and activity is very low. Transcription of these enzymes is tightly regulated by cytokines or growth factors, including transforming growth factors, interleukins (I -1, I -4, I -6) or tumor necrosis factor alpha(TNFα) (Sternlicht and Werb, 2001) . Post-transcriptionally, MMP activity is controlled by interaction between zinc-containing catalytic site and N-terminal pro-peptide domain.
When this balance between MMPs and their natural inhibitors is shifted towards enzyme expression and activity, increased tissue degradation occurs. Increased levels of MMP expression have been shown to be involved in a large number of pathological conditions, such as arthritis, Alzheimer’s disease, cardiovascular disease, as well as cancer. MMPs have now been therefore considered important pharmaceutical targets and extensive effort have been put into de-sign of potential drugs based on MMP inhibition.
The active site of MMPs features two distinct regions. First one is a groove centered on the catalytic zinc. Second one is a S1’ specificity site, which varies among different members of a family and is important for substrate (or inhibitor) selectivity. The volume of thisS1’ subsite varies highly from a small hydrophobic pocket of MMP-7 to a very large site in MMP-8.
The strength of interaction between receptor protein and ligand is very important (but not the only one) criterion to distinguish potent inhibitors (i.e. potential drugs) from the non-binding compounds. This attractive force between protein and ligand is called binding affinity. It is influenced by non-covalent interactions, such as hydrogen bonding, electrostatic or van der Waals interactions. As mentioned earlier, MMPs are promising pharmaceutical targets, especially for cancer therapy. arge number of both synthetic and natural inhibitors have been identified and tested in clinical trials, but so far with only limited success. While many of these compounds showed cytostatic or anti-angiogenic activity, discovered side effects or low specificity leading to excessive inhibition of MMPs not involved in the particular pathological process led to disappointing results. Current effort is now focused on computer-aided design of more specific inhibitors based on knowledge of three-dimensional structure of many MMPs.
The requirements for a compound to be a potent MMP inhibitor are following: a) a functional group capable of chelation of catalytic zinc(II) ion [e.g. carboxylate (COO-), thiolate (S-) or hydroxamate (CONH-O-)]; b) one or more functional groups capable of interacting with enzyme backbone via hydrogen bonds; c) at least one functional group, which will undergo van der Waals interactions with the protein subsites. The present study has been undertaken to look simulation of selected target through phytochemical calendoflavobioside of Calendula officinalis inn.(Asteraceae)
MATERIALS AND METHODS
Protein preparation
AutoDock is a suite of automated docking tool. It is designed to predict how small molecules, such as substrates or drug candidates, bind to a receptor of known 3D structure (Morris et al ., 1998) . The protein add with Kollmann charges were assigned. Through which hydrogens were added, side chains were optimized for hydrogen bonding. The energy minimized protein was then saved in PDB format. Using MG Tools-1.4.6 nonpolar hydrogens were merged, AutoDock atom type AD4 and Gasteiger charges were assigned and finally saved in protein.pdbqt format (Morris et al ., 1998).
Ligand preparation
Structure of ligands were drawn using ChemSketch, optimized with 3D-geometry and the two-dimensional structure of plant compound are converted into 3-D structure using the Babel format molecule converter (Guha et al ., 2006) and saved in PDB format for AutoDock compatibility. MG Tools-1.4.6 (The Sripps Research Institute) was used to convert ligand.pdb files to ligand.pdbqt files.
Docking protocol
Grid parameter files (protein.gpf) and docking parameter files (ligand.dpf) have written using MG Tools-1.4.6. Receptor grids were generated using 90x60x60 grid points in xyz with grid spacing of 0.375Å. Grid box was centered co crystallized ligand map types were generated using autogrid. Docking of macromolecule was performed using an empirical free energy function and amarckian Genetic Algorithm, with an initial population of 250 randomly placed individuals, a maximum number of 106 energy evaluations, a mutation rate of 0.02, and a crossover rate of 0.80. One hundred independent docking runs were performed for each ligand. Results differing by 2.0Å in positional rootmean square deviation (RMSD) were clustered together and represented by the result with the most favorable free energy of binding . It is expressed in the units-kcal/mol.
RESULTS AND DISCUSSION
Docking studies of a new compound from Calendula officinalis Linn.:
Ligand Selection
In silico study has approached to support the mechanism of action of the phyto-compounds against target protein in wound healing. With the help of spectra, compounds in selected plants were identified and made attempt for docking against of MMP 8 and MMP 12 to prove for its wound healing potentials.
The ligand Calendoflavobioside from selected plant were selected for docking analysis.
Target Selection
MMPs are proteolytic and matrix degrading enzymes play multiactions in the pathology of the nervous system. Among the various factors, the extracellular matrix (ECM) made from fibrin, elastin, hyaluronan, type I and type III collagen are combined together to form a fibrenetwork to maintain the structural integrity (Gill and Parks, 2008). MMP contained different class of enzymes, especially collagenase (MMP8) and metalloelastase (MMP12) involved in the tissue repairing process (Sternlicht and Werb, 2001).
The human matrix metalloproteinases
(MMPs)comprise a family of secreted and transmembrane zinc-dependent endopeptidases that degrade the macromolecular components of the extracellular matrices (ECM) and basement membrane components. Under normal physiological conditions, the proteolytic activities of enzymes are controlled by tissue inhibitors of matrix metalloproteinases. In pathological conditions, this balance is shifted towards overactivation of MMPs leading to excessive degradation of the matrix components. ExcessiveMMP activity has been implicated in numerous disease states involving matrix degradation, which include arthritis, periodontal diseases,osteogenesis imperfecta, Alzheimer’s disease and tumor invasion and metastasis (Sobhani et al ., 2004).
Currently, at least 22 structurally related zinc dependent members with a broad spectrum of proteolytic activity against several components of the ECM have been identified. Three-dimensional (3D) structures of many of these MMPs (MMP-1 to MMP-3, MMP7 to MMP-9 and MMP-11 to MMP-13) have been determined by the X-ray crystallography andNMR techniques. There are more than sixty 3Dstructure entries related to MMPs in Protein DataBank (PDB) of which more than half of themare in complex with various small molecularinhibitors. MMP inhibitors (MMPIs) are consistedof a zinc binding group (ZBG), including but notlimited to hydroxamates (the most potent),carboxylates, phosphinates, and thiolates, coupledto a framework with varying numbers of pocketoccupying side chains. Availability of this relatively high number of 3D structures incomplex with different inhibitors provides a great opportunity for docking studies (Park et al ., 2014).
Docking studies
The docking poses were ranked according to their docking scores, list of docked ligands and their corresponding binding poses. After the simulations were complete, the docked structures were analyzed and the interactions were observed. Hydrogen bond interactions and binding distance between the donors and acceptors were measured for the best conformers. Distinct conformation clusters RMSD (Root Mean Square
Deviation) -tolerance and Van der Waals scaling factor were found to be 2.0 Å. 1.0 Å respectively.
Molecular docking studies were performed by docking Matrix Metalloproteinase 8(3DNG) and Matrix Metalloproteinase 12 (5 AB) with Calendoflavobioside and the binding energy was calculated of these complex structure. The compound showed negative binding energy with the both target proteins MMP 8 and MMP12. The negative energy has excellent binding to the receptor compound. Docking the target protein with plant metabolite can be performed by docking and binding energy was showed in Table 3 Free energy of binding is calculated as a sum of four energy terms of intermolecular energy (van der waals, hydrogen bond, desolvation energy and electrostatic energy), total internal energy, torsional free energy and unbound system energy.
The amino acid interactions in the target protein binding site and important H-bonds with residues: G U108, A A163, ASN190, EU193, HIS201, HIS207, TYR216, PRO217, TYR219, A A220, ARG222, TYR227 andSER228 of MMP8 (Table 4) and EU181, A A182, A A184, G U219, HIS222, HIS228; YS241 and PHE237 of MMP12 (Table 5). The binding energies of the target protein-ligand interactions are important to describe that how the lignad binds to the target. These results suggested that calendoflavobioside has very high affinity for binding site of target proteins and probably act as competitive inhibitor as shown in Table 3,4 and 5.Inhibition of matrix metalloproteinase might be a valuable approach for the treatment of chronic diseases. Collagenase are zinc dependent endopeptidase which causes bronchitis inflammation in between the nose and lungs (Lemjabbar et al., 1999). It is also a novel modulator of inflammation during sepsis.
MMP-8 has a stronger affinity toward type I collagen and involved in various inflammatory processes. Docking studies revealed that calendoflavobioside may decrease collagenase activity by binding with MMP-8, which in turn increases the collagen 1 content of the extracellular matrix A major substrate for MMP-12 is elastin, but MMP-12 is capable of degrading other ECM constituents. Elevated MMP-12 levels have been measured in various diseases. By inhibiting elastase, calendoflavobioside may increase the amount of elastin which is important for wound strength. Excessive and prolonged expression and activation of MMPs are etiologic causes of chronic diseases due, at least in part, to excessive tissue destruction . The result made in present study showed that calendoflavobioside can bind with MMP 8 and MMP-12 may decrease their activity.
Table 1. Compounds ( ipinskiRule of Five) –Details taken from Pubchem compound
|
S.No |
Compound |
PubChem CID |
Molecular formula |
XLog P |
Molecular weight (g/mol) |
Hydrogen bond donor |
Hydrogen bond acceptor |
Rotatio nal bonds |
|
1 |
Calendoflavobioside |
5491657 |
C 27 H 30 O 16 |
-0.7 |
610.521 g/mol |
10 |
16 |
6 |
Table - 2. Protein Details from PDB
|
S.No |
Protein Name |
PDB ID |
Experiment |
Resolution |
Protein bound Inhibitor |
|
1 |
Metalloproteinase 8 |
3DNG |
X-ray |
2.0Å |
5S)-5-(2-amino-2-oxoethyl)-4-oxo-N-[(3-oxo- 3,4- dihydro-2H-1,4-benzoxazin-6-yl)methyl]- 3,4,5,6,7,8- hexahydro[1]benzothieno[2,3-d]pyrimidine- 2-carboxamide |
|
2 |
Metalloproteinase 12 |
5 AB |
X-ray |
1.34Å |
n-isobutyl-n-[4-methoxyphenylsulfonyl]glycylhy droxamic acid |
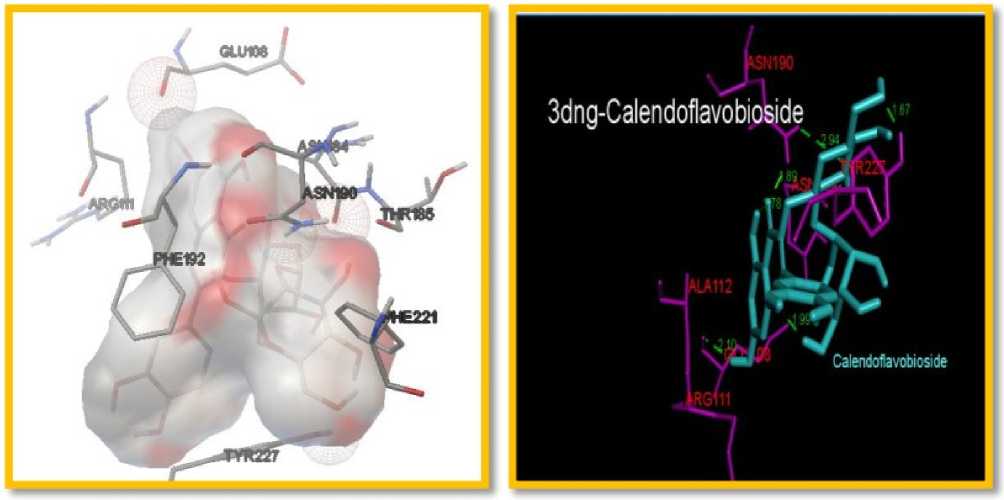
Figure 1. The docking interaction of Metalloproteinase 8 (3DNG) with Selected igands (Calendoflavobioside
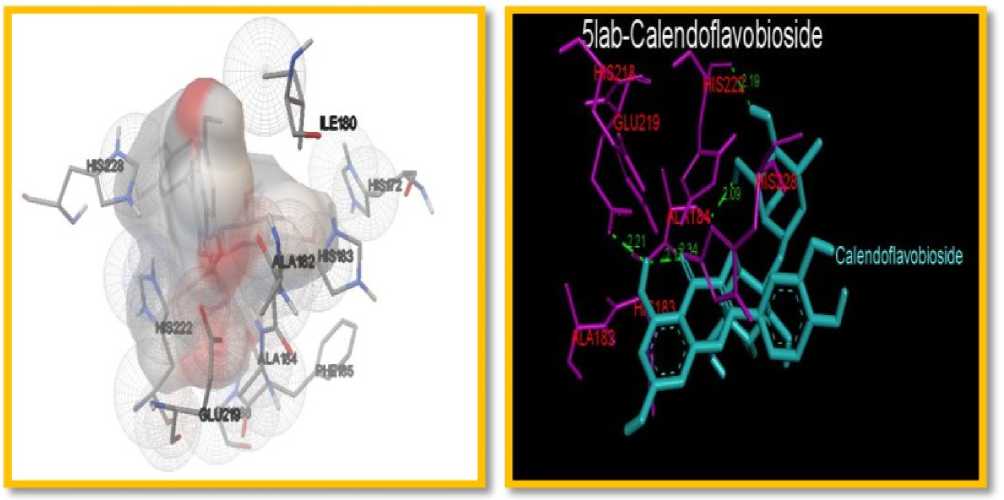
Figure 2. The docking interaction of Metalloproteinase12(5 AB with Selected igands (Calendoflavobioside
Table 3. Docking Parameters: Molecular Docking Result of MMP 8 (3dng
|
S.No |
Protein – Ligand Complex |
Binding Energy (Kcal/mol) |
Ligand Efficiency (kcal/mol) |
Ki value ( µM) |
No of Favourable Hydrogen Bonds |
|
3DNG- (Metalloproteinase 8) |
|||||
|
1 |
3DNG-Calendoflavobioside |
-3.0 |
-0.07 |
6.29mM |
4 |
|
5LAB-(Metalloproteinase 12) |
|||||
|
1 |
5 AB-Calendoflavobioside |
-3.08 |
-0.07 |
5.49 |
4 |
Table 4. Hydrogen bonding patterns of MMP 8
|
Ligands |
Distance (Å) |
H-bonding |
|
3DNG-METALLOPROTEINASE 8 (MMP8) |
||
|
3DNG-Calendoflavobioside |
2.94 |
A:ASN190:HD21 - :Calendoflavobioside:O5 |
|
1.89 |
A:ASN190:HD22 - :Calendoflavobioside:O12 |
|
|
2.10 |
:Calendoflavobioside:H71 - A:G U108:O |
|
|
1.67 |
A:TYR227:HH - :Calendoflavobioside:O10 |
|
Table 5:Hydrogen bonding patterns of MMP 12
|
Ligands |
Distance (Å) |
H-bonding |
|
5LAB-METALLOPROTEINASE 12 (MMP12) |
||
|
5 AB-Calendoflavobioside |
2.21 |
:Calendoflavobioside:H70 - A:G U219:OE1 |
|
2.34 |
A:A A184:HN - :Calendoflavobioside:O12 |
|
|
2.09 |
:Calendoflavobioside:H62 - A:A A184:O |
|
|
2.19 |
:Calendoflavobioside:H63 - A:HIS222:O |
|
CONCLUSION
The findings of in silico research indicate that calendoflavobioside may help promote wound healing through its interaction with the process's target proteins, MMP 8 and MMP 12. To validate the efficacy of these plant compounds in accelerating wound healing, more in vitro and in vivo research is required.
CONFLICTS OF INTEREST
The authors declare that they have no potential conflicts of interest.
