Инструментальные методы определения степени деацетилирования хитина

Автор: Кучина Юлия Анатольевна, Долгопятова Наталия Владимировна, Новиков Виталий Юрьевич, Сагайдачный Вадим Александрович, Морозов Николай Николаевич
Журнал: Вестник Мурманского государственного технического университета @vestnik-mstu
Статья в выпуске: 1 т.15, 2012 года.
Бесплатный доступ
Приведен обзор методов определения степени деацетилирования хитина. Определена оптическая плотность образцов хитозана с различной молекулярной массой в инфракрасной области спектра. Приведены сравнительные результаты определения степени деацетилирования хитина методами инфракрасной спектроскопии и с помощью регистрации продуктов кислотного гидролиза хитина методом жидкостной хроматографии.
Хитин, хитозан, кислотный гидролиз, степень деацетилирования, молекулярная масса
Короткий адрес: https://sciup.org/14294411
IDR: 14294411
Текст научной статьи Инструментальные методы определения степени деацетилирования хитина
Разработка комплексных малоотходных технологий переработки морских гидробионтов, в частности промысловых ракообразных, составляет одну из важных задач современного развития рыбохозяйственного комплекса.
Основным продуктом утилизации больших объемов отходов переработки ракообразных является хитин, который вследствие плохой растворимости не находит широкого применения. Химический гидролиз хитина (кислотный и щелочной) представляет собой важный метод модификации этого природного полисахарида с целью получения хитозана, хитоолигосахаридов и мономеров ( Орлова и др. , 1989).
Исторически термины "хитин" и "хитозан" сложились, исходя из главного различия свойств этих полисахаридов, а именно растворимости в разбавленных кислотах. Растворимость этих полисахаридов в слабых и разбавленных кислотах определяется степенью деацетилирования, т.е. содержанием в молекуле свободных аминогрупп. Когда степень деацетилирования менее 50 %, биополимер принято называть хитином. При степени деацетилирования более 50 % (обычно более 60 %) полисахарид становится растворимым в разбавленных растворах органических и неорганических кислот. Такой полисахарид получил название хитозан. Обычно этот термин используется для глубоко деацетилированного хитина, который является растворимым в разбавленной уксусной кислоте ( Быкова, Немцев , 2002; Ottoy et al. , 1996).
Хитин – линейный аминополисахарид, состоящий из N-ацетил-2-амино-2-дезокиси-D-гликопиранозных звеньев.
Структурная формула хитина представлена на рис. 1.
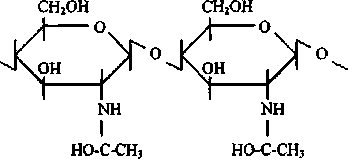
Рис. 1. Структурная формула "идеального" хитина
Хитозан образуется путем N-деацетилирования хитиновой молекулы. Доля деацетилированных единиц, определяемая как степень деацетилирования (DD), составляет обычно от 70 до 90 %. Эти параметры могут изменяться в зависимости от источника хитина и методов технологической обработки. Структурная формула частично деацетилированного хитина представлена на рис. 2.
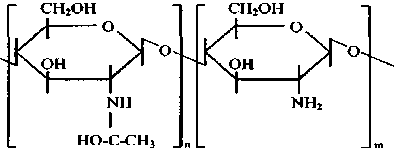
Рис. 2. Структурная формула частично деацетилированного хитина
Степень ацетилирования (DA) хитина (или хитозана) определяется как отношение количества N-ацетилглюкозаминных звеньев ( n ) к общему числу мономерных звеньев в молекуле полисахарида:
Da = n / ( n + m ). (1)
Степень деацетилирования (DD) - это отношение количества глюкозаминных звеньев ( m ) к общему числу мономерных звеньев в молекуле полисахарида:
DD (%) = n / ( n + m ) = N а MИHHЫЙ / N общий = 1 - DA. (2)
Крайние состояния представляют собой полностью ацетилированные и полностью деацетилированные полисахариды, которые можно считать идеальными хитином и хитозаном. Обычно в природе не существует полностью ацетилированного или деацетилированного полисахарида.
Хитин и хитозан, являясь полисахаридами переменного состава, представляют собой смесь полимерных молекул, отличающихся по молекулярной массе, по степени деацетилирования, по распределению однотипных звеньев внутри молекулы.
В процессе щелочного гидролиза хитина протекает главным образом реакция деацетилирования. В процессе кислотного гидролиза хитина параллельно с реакцией гидролиза гликозидных связей, приводящей к деструкции и деполимеризации, протекает реакция деацетилирования. На протекание этих реакций могут оказывать влияние такие физико-химические свойства хитина и хитозана, как степень деацетилирования и молекулярная масса.
Поскольку степень деацетилирования является важной характеристикой хитина и хитозана, определяющей их свойства и области применения, остановимся на основных способах ее определения.
Степень деацетилирования можно определить методом инфракрасной (ИК)-спектроскопии ( Domszy, Roberts , 1985), потенциометрического титрования ( Broussignac , 1968), кислотным гидролизом с последующим определением уксусной кислоты хроматографическими методами ( Niola et al. , 1993), ферментативным гидролизом ( Nanjo et al. , 1991), методом ультрафиолетовой (УФ)-спектроскопии, спектроскопией ядерного магнитного резонанса ( Saito et al. , 1987; Varum et al. , 1991), методом кругового дихроизма ( Domard , 1987) и элементным анализом. Некоторые из этих методов не подходят для нерастворимых или малорастворимых образцов хитина и хитозана. В других методах образцы должны быть тщательно очищены для удаления нерастворимой фракции, некоторые из них дороги, характеризуются невысокой точностью, длительны при выполнении.
Целью работы является обзор известных и наиболее доступных методов определения степени деацетилирования хитина и хитозана; определение оптической плотности образцов хитозана с различной молекулярной массой в инфракрасной области спектра; сравнение результатов определения степени деацетилирования хитина методом инфракрасной спектроскопии и с помощью кислотного гидролиза хитина с последующей регистрацией продуктов гидролиза методом высокоэффективной жидкостной хроматографии.
-
2. Методы определения степени деацетилирования хитина
-
2.1. Метод элементного анализа
-
2.2. Метод потенциометрического титрования
Наиболее доступными являются методы ИК-спектроскопии, потенциометрического титрования, кислотного гидролиза и элементного анализа. Каждый из этих методов имеет свои достоинства и недостатки. Так, например, методом потенциометрического титрования можно определять DD только растворимых образцов хитозана; для определения степени деацетилирования методом ИК-спектроскопии необходимы стандартные образцы с известной DD. Поэтому использование различных методов для характеристики степени деацетилирования хитина и хитозана позволяет оценить этот показатель с наибольшей достоверностью.
Элементным анализом степень деацетилирования определяют как процент азота, рассчитанный по органической фракции анализируемого полисахарида. Анализируемый образец сжигают при температуре 600 °С, долю неорганического вещества определяют взвешиванием остатка после сжигания, степень деацетилирования рассчитывают по формуле:
DD (%) = [(8,695 – N) / 1,799] × 100, (3)
где 8,695 – процент азота в полностью деацетилированном хитозане; 1,799 – разница между 8,695 и 6,896 (процент в полностью ацетилированном хитине); N (%) – процент азота, рассчитанный по органической фракции анализируемого материала.
Элементный анализ характеризуется невысокой воспроизводимостью результатов.
Метод основан на потенциометрическом титровании хлористого водорода, связанного с аминогруппами молекул хитозана.
Потенциометрическое титрование растворов хитозана раствором гидроксида натрия точной концентрации проводили при помощи иономера Анион 4151 (НПП "Инфраспак-Аналит", Россия) с шагом 0,1 см3. Полученную кривую (зависимость рН раствора от объема титранта) графически обрабатывали и находили объемы титранта (NaOH), соответствующие точкам эквивалентности соляной кислоты и солянокислого хитозана.
Степень деацетилирования образца (DD), в процентах, вычисляли по формуле:
DD = 203,2 × 100 / [42,0 + 1000 m 0 / ( С NaOH dV NaOH )], (4) где m 0 – масса хитозана в навеске, г; С NaOH – точная концентрация раствора гидроксида натрия, моль/дм3; dV NaOH – объём раствора гидроксида натрия, пошедший на титрование аминогрупп, смЗ; 203,2; 42,0; 100; 1000 – пересчётные коэффициенты.
За окончательный результат принимали среднее арифметическое значение результатов трёх параллельных измерений, относительное расхождение между которыми не превышало 2 %.
В данной работе метод потенциометрического титрования применяли для определения DD образцов хитозанов, которые далее использовали для построения калибровочного графика при определении степени деацетилирования методом инфракрасной спектроскопии.
-
2.3. Метод инфракрасной спектроскопии
Инфракрасная спектроскопия (в таблетках KBr или пленках) широко используется для определения степени деацетилирования хитина и хитозана ( Domszy et al. , 1985; Khan et al. , 2002; Wang et al. , 2004).
Спектры поглощения в ИК-области записывали на инфракрасном спектрофотометре IR-420 (Shimadzu, Япония) в диапазоне частот от 4000 до 400 см-1. Анализ проводили в лаборатории ФГБУП "Полярный научно-исследовательский институт рыбного хозяйства и океанографии им. Н.М. Книповича" (ПИНРО).
В работе использовали крабовый хитин, полученный по технологии ( Долгопятова , 2011). Для приготовления образцов с различной степенью деацетилирования и молекулярной массой проводили кислотный гидролиз хитина в растворе хлороводородной кислоты с концентрацией 1,0 моль/дм3 при температуре от 50 C.
Молекулярную массу (ММ) образцов, полученных в процессе кислотного гидролиза, определяли вискозиметрически ( Павлов, Селюнин , 1986). Гидролизованные образцы деацетилировали в стандартных условиях. Стандартное деацетилирование гидролизованных в НСl образцов хитина проводили в 50%-ном растворе NaOH при 100 C.
В работе Чеботок и др. (2006) было установлено, что в условиях стандартного деацетилирования степень полимеризации хитина и хитозана практически не изменяется.
Исследуемые образцы измельчали на мельнице TI-100 (C.M.T. Co., Ltd., Япония) в течение 15 мин и доводили до постоянной массы высушиванием при 100 C. KBr предварительно перекристаллизовывали из водного раствора и прокаливали при 600 C. Качество KBr проверяли по инфракрасному поглощению. Для приготовления таблеток использовали смесь образца и KBr в соотношении 1:80 по массе. Таблетки готовили из навески 120 мг смеси (диаметр и толщина таблеток составляли соответственно 13 и 0,42 мм), прессовали под давлением 650 кгс/см2 в течение 1 мин при комнатной температуре.
На образцах хитина и хитозана с DD от 0 до 100 % ранее было установлено ( van Scheltinga , 1995), что наиболее подходящей для измерения опорной частотой является 2878 см-1. Поглощение на этой частоте обусловлено валентными колебаниями связей C-H, и величина оптической плотности сохраняется постоянной для образцов с разной степенью деацетилирования. Деформационным колебаниям аминогруппы (Амид I) соответствует частота 1655 см-1. Пример обработки ИК спектра приведен на рис. 3.
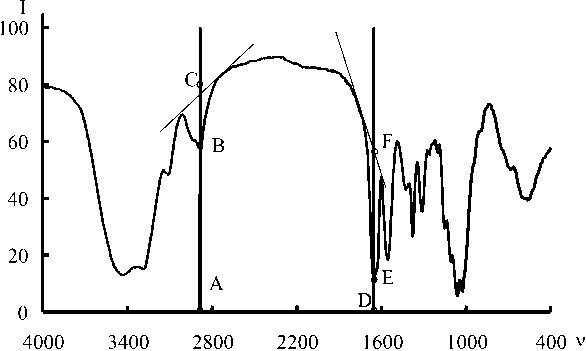
Рис. 3. ИК-спектр поглощения хитина в таблетке KBr. Линии и точки показывают пример графической обработки данных для расчета DD. I – пропускание, %; V – частота излучения, см-1
Оптическую плотность рассчитывали по формулам:
A 2878 = lg(AC/AB),
A 1655 = lg(DF/DE).
Степень деацетилирования образцов определяли по калибровочному графику, полученному для образцов хитозана с известным значением DD (рис. 4). Степень деацетилирования образцов хитозана, используемых для построения калибровочного графика, определяли методом потенциометрического титрования.
A 1655 /A 2878
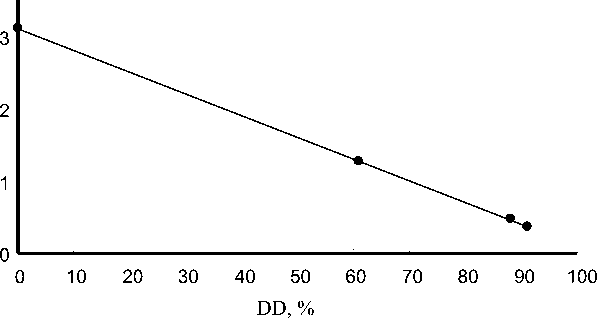
Рис. 4. Зависимость поглощения хитина/хитозана в ИК-области от степени деацетилирования. Таблетка KBr. A1655/A2878 – отношение оптической плотности на частоте 1655 см-1 к оптической плотности на опорной частоте 2878 см-1; DD – степень деацетилирования, %
Полученное уравнение регрессии имеет вид:
А1655/А2878 = –0,03 X DD + 3,1078, ( r 2 = 0,9992).
Результаты изучения зависимости относительной оптической плотности образцов хитозана с различной молекулярной массой и степенью деацетилирования приведены на рис. 5. А = A1655/A2878 – относительная оптическая плотность.
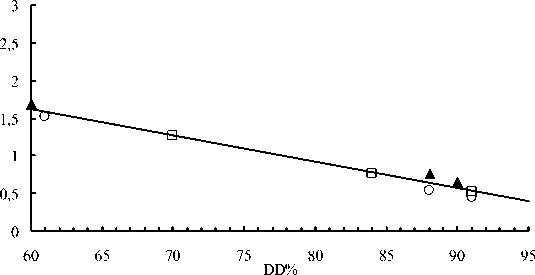
Рис. 5. Зависимость относительной оптической плотности образцов хитозана с различной молекулярной массой от степени деацетилирования. ММ = 510650 Да (▲), 50270 Да (□) и 10650 (○)
Из рис. 5 следует, что относительные оптические плотности образцов хитозана с одинаковой DD, но различной ММ практически совпадают. Из полученных результатов можно сделать заключение о том, что калибровочной зависимостью А – DD, приведенной на рис. 5, можно пользоваться для определения степени деацетилирования образцов, характеризующихся различной молекулярной массой.
К недостаткам определения степени деацетилирования методом ИК-спектроскопии можно отнести необходимость определения степени деацетилирования образцов, используемых для построения калибровочной зависимости, другими методами. Чаще всего для построения калибровочного графика используют растворимые образцы хитозанов, DD которых определяется методом потенциометрического титрования. Полученную для образцов хитозана зависимость А – DD экстраполируют до нулевых значений DD.
-
2.4. Метод кислотного гидролиза
Перспективным методом для определения степени деацетилирования хитина в диапазоне DD от 0 до 100 % является кислотный гидролиз полисахарида с последующим количественным определением выделившейся в процессе деацетилирования уксусной кислоты. Впервые этот метод был использован для определения ацетильных групп в древесине и сопутствующих изделиях ( Niola et al. , 1993). В работе Niola et al. (1993) гидролиз хитина проводили в присутствии серной и щавелевой кислот, что увеличивает эффективность процесса гидролиза, т.к. серная кислота частично деполимеризует полисахарид, снижая его молекулярную массу, а далее щавелевая кислота деацетилирует. Выделяющуюся при деацетилировании хитина и хитозана уксусную кислоту авторы этой работы определяли спектрофотометрически на λ = 210-215 нм после разделения реакционной смеси методом жидкостной хроматографии. Этот метод применим для полного диапазона степеней ацетилирования, результаты определения DD совпадают с результатами, полученными другими методами. Метод кислотного гидролиза характеризуется непродолжительным временем анализа по сравнению с другими методами.
Для определения степени деацетилирования методом кислотного гидролиза в данной работе использовали образцы крабового хитина. Для приготовления образцов с различной степенью деацетилирования проводили гидролиз хитина в 1 н хлороводородной кислоте (НСl) в течение 5, 10, 20, 40, 50 и 60 мин. Полученные таким образом образцы далее обрабатывали смесью двух кислот (0,5 см3 12 М H2SO4 и 2,0 см3 0,0063 % раствора дигидрата щавелевой кислоты) в запаянных ампулах при температуре 155 °С в течение 1 ч.
Для хроматографического анализа гидролизованные в ампулах в присутствии серной и щавелевой кислот образцы хитина готовили следующим образом: ампулы разбивали, их содержимое количественно переносили в колбу, нейтрализовали избыток кислоты раствором Ва(ОН) 2 до рН = 2,0-2,5, доводили объем раствора до 50 см3, удаляли образовавшийся нерастворимый сульфат бария центрифугированием (на центрифуге ОПН-8 при скорости 8000 об/мин в течение 20 мин). Разделение продуктов гидролиза хитина после обработки смесью кислот (серной и щавелевой) и количество выделившейся уксусной кислоты определяли методом жидкостной хроматографии на приборе "Милихром-4".
Перед введением исследуемого образца в хроматографическую колонку его предварительно фильтровали через мембрану из нержавеющей стали с размером пор 0,45 мкм.
Разделение продуктов гидролиза проводили на колонке Nucleosil C-18 (150X4 мм, 5 мкм). Элюент – бидистиллированная вода с добавлением 0,05 % орто-фосфорной кислоты, скорость подачи 40 мкл/мин, объем вводимой пробы – 10 мкл. Выходящие фракции регистрировали с помощью УФ-детектора на длине волны 214 нм.
На рис. 6 в качестве примера приведены хроматограммы образцов хитина (время гидролиза образцов в НСl 5 и 10 мин). Для других исследованных образцов получены аналогичные результаты.
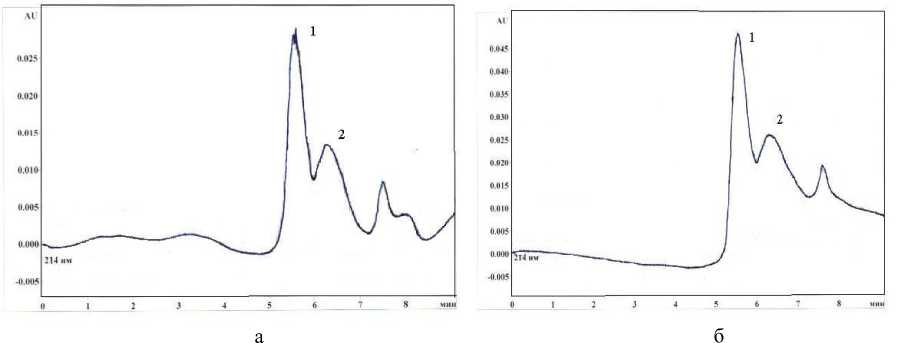
Рис. 6. Хроматограммы образцов хитина: а – время гидролиза 10 мин, б – время гидролиза 5 мин.
Пик 1 – щавелевая кислота, пик 2 – уксусная кислота
Как видно из хроматограмм, в условиях эксперимента получено разделение хроматографических пиков уксусной и щавелевой кислот. Степень деацетилирования определяли по количеству выделившейся уксусной кислоты по формуле ( Niola et al. , 1993):
DD (%) = [161 X / (43 – 42 X )] × 100, (8)
где 161 – молекулярная масса звена 2-амино-2-деокси-D-глюкозы; 43 – молекулярная масса ацетильной группы; Х = mх / М ( mх – количество выделившейся уксусной кислоты; М – количество полисахарида, взятого на анализ).
Результаты определения степени деацетилирования хитина кислотным гидролизом, с последующим разделением продуктов гидролиза методом жидкостной хроматографии приведены в табл. 1.
Таблица 1. Степень деацетилирования хитина, определенная кислотным гидролизом, с последующим разделением продуктов гидролиза методом жидкостной хроматографии
|
Время гидролиза, мин |
— |
5 |
10 |
20 |
|
DD, % |
1,02 |
4,4 |
8,8 |
16,1 |
В табл. 2 приведены сравнительные результаты определения степени деацетилирования хитина методом ИК-спектроскопии и методом кислотного гидролиза с последующим разделением продуктов гидролиза методом жидкостной хроматографии. Для приготовления образцов с различной степенью деацетилирования проводили гидролиз хитина в 1 н НСl в течение 40-60 мин.
Таблица 2. Степень деацетилирования хитина, определенная методом ИК-спектроскопии и методом кислотного гидролиза
|
Время гидролиза, мин |
0 |
40 |
50 |
60 |
|
DD, % (метод кислотного гидролиза) |
1,02 |
39 |
46 |
48 |
|
DD, % (метод ИК-спектроскопии) |
1,2 |
43 |
50 |
51 |
Полученные результаты определения DD методом кислотного гидролиза с последующим разделением продуктов гидролиза с помощью жидкостной хроматографии хорошо согласуются с данными по определению степени деацетилирования в образцах хитина методом ИК-спектроскопии.
-
3. Заключение
-
1. Показано, что относительные оптические плотности образцов хитозана в инфракрасной области спектра с одинаковой степенью деацетилирования, но различной молекулярной массой практически совпадают.
-
2. Значения степени деацетилирования образцов хитина, полученные методом кислотного гидролиза с последующим разделением продуктов с помощью жидкостной хроматографии, хорошо согласуются в данными по определению степени деацетилирования, полученными методом ИК-спектроскопии.
Научно-исследовательская работа выполнена в рамках реализации ФЦП "Научные и научнопедагогические кадры инновационной России" на 2009-2013 гг.
