Интегрированный подход к ведéнию детей с генетическими нарушениями на примере ребенка с синдромом Аперта (клинический случай)
")
Автор: Глушаков И.А., Гуменюк О.И., Тяпкина Д.А., Гаджикеримов Г.Э., Черненков Ю.В.
Журнал: Саратовский научно-медицинский журнал @ssmj
Рубрика: Педиатрия
Статья в выпуске: 3 т.21, 2025 года.
Бесплатный доступ
В статье приведен клинический случай с синдромом Аперта у ребенка 2 лет. Синдром Аперта является аутосомно-доминантным наследственным краниосиностозом, развивающимся вследствие патогенного варианта гена FGFR2, что приводит к дефициту внутриклеточных сигналов, которые регулируют эмбриогенез, вызывая преждевременную гаструляцию, аномалии имплантации, нарушение эпителиальномезенхимальных взаимодействий, обусловливая тяжесть состояния пациентов. В статье продемонстрирован комплексный подход к ведéнию и наблюдению такого пациента с привлечением специалистов различного профиля.
Синдром Аперта, фенотипические особенности, репродуктивный статус, фактор роста фибробластов 2, краниосиностоз
Короткий адрес: https://sciup.org/149149430
IDR: 149149430 | УДК: 616-002.5-053.2-084:615.28 | DOI: 10.15275/ssmj2103301
Integrated approach to care of children with genetic disorders on the example of a child with Apert syndrome (clinical case)
The article presents a clinical case of Apert syndrome in a 2-year-old child. Apert syndrome is an autosomal dominant hereditary craniosynostosis that develops due to the pathogenic variant of the FGFR2 gene, which leads to a deficiency of intracellular signals that regulate embryogenesis, causing premature gastrulation, implantation abnormalities, violation of epithelial-mesenchymal interactions, which determines the severity of the patient’s condition. The article demonstrates an integrated approach to the management and observation of such a patient with the involvement of specialists of various profiles.
Текст научной статьи Интегрированный подход к ведéнию детей с генетическими нарушениями на примере ребенка с синдромом Аперта (клинический случай)
EDN: LTKQWW
-
1Введение. Акроцефалосиндактилия I типа, или синдром Аперта, – это редкая форма акроцефало-синдактилии, которая представляет собой врожденную патологию, характеризующуюся краниосиностозом, гипоплазией лицевого черепа и синдактилией конечностей. Синдром назван в честь французского педиатра Эжена Аперта, который впервые в 1906 г. описал 9 человек с подобными характеристиками [1, 2].
Дети с синдромом Аперта могут сталкиваться с рядом трудностей, связанных с нормальным развитием. Важно учитывать возможность возникновения сопутствующих заболеваний и осложнений, которые могут оказывать влияние на репродуктивную функцию и, как следствие, осложнять процесс взросления и полового созревания детей с таким синдромом. Последний является редким заболеванием и, по оценкам экспертов, встречается у 1 на 65 тыс. новорожденных. Синдром Аперта относится к одному из 4 типов краниосиностоза и составляет примерно 4,5% от всех случаев преждевременного сращения черепных швов: приблизительно 15 на 1 млн новорожденных. Риск развития заболевания значительно увеличивается с возрастом отца и, как считается, обеспечивает селективное преимущество в мужских сперматогониальных клетках [3].
Синдром Аперта имеет аутосомно-доминантный тип наследования с полной пенетрантностью, но вариабельной экспрессией (в большинстве случаев он возникает спонтанно вследствие новых мутаций и зародышевого мозаицизма). Однако патогенез редких случаев рецидива у здоровых родителей до сих пор остается неясным.
Заболевание развивается в результате миссенс-му-тации FGFR2 (fibroblast growth factor receptor 2) – гена рецептора фактора роста фибробластов 2 в хромосоме 10q26, с заменой аминокислот S252W или P253R.10q. В настоящее время в медицинской литературе зарегистрировано более 300 случаев патогенного варианта FGFR2 [4–6].
Клинические особенности данного синдрома включают следующие фенотипические проявления: краниосиностоз, гипоплазия лицевого черепа и симметричная синдактилия нижних и верхних конечностей [6]. Признаки гипертелоризма (широко расставленные глаза), проптозиса (выпученные глаза) и косых глазных трещин являются чертами лица, обнаруженными в некоторых краниосиностозах. Существуют и другие деформации лицевого черепа, обнаруженные при синдроме Аперта и других краниосиностозах, такие как акроцефалия (конусообразный череп), проптоз, выступающий лоб, гипертелоризм, наклоненные вниз глазные щели и уплощенная спинка носа. Зубочелюстные поражения включали скученность зубов, высокое дугообразное нёбо, узкое нёбо и псевдорасщелины. Могут наблюдаться патологии со стороны внутренних органов и другие аномалии скелета, такие как шейный спондилодез. Возможна также умственная отсталость легкой и умеренной степени [4]. Особенности строения кисти – короткий, радиально искривленный 1-й палец, сложная синдактилия 2, 3 и 4-й фаланг пальцев и симфалангизм (врожденная ригидность пальцев из-за неспособности кости полностью отделиться, как это обычно происходит во время внутриутробного развития плода).
Цель – на приеме клинического случая представить сложности диагностики, лечения и наблюдения ребенка с синдромом Аперта.
Информирование согласие от законных представителей пациентки на публикацию данных из истории болезни получено.
Описание клинического случая. Пациентка Х. 2 лет с синдромом Аперта. Жалобы (со слов матери) на низкий рост и плохую прибавку в весе. При оценке анамнеза определено, что ребенок со множественными врожденными порокам развития: черепно-лицевым дисморфизмом, неполной расщелиной твердого и мягкого нёба, полидактилией обеих стоп, тотальной синдактилией всех конечностей. Синдром Аперта установлен клинически. Девочка от I беременности на фоне эрозии шейки матки, I срочных родов, тазового предлежания плода. Семейный анамнез – без особенностей. Оценка по шкале Апгар – 6/7 баллов. Девочка родилась с массой тела 3110 г, длиной – 54 см, окружностью головы – 37 см, окружностью груди – 32 см. С рождения состояние средней степени тяжести. Энтеральное питание молоком через назогастральный зонд. Привита вакциной туберкулезной для щадящей первичной иммунизации (БЦЖ-М). Профилактические прививки – согласно национальному календарю.
При осмотре оценено физическое развитие ребенка: рост – 79,5 см (SDS -2,63 с.о.), масса тела – 8,8 кг, индекс массы тела – 13,9 кг/м2 (SDS -1,39 с.о.), допубертатное половое развитие. Из особенностей фенотипа: низкорослость, черепно-лицевой дисморфизм («кукольное» лицо), неполная расщелина твердого и мягкого нёба, полидактилия обеих стоп, тотальная синдактилия всех конечностей (рисунок).
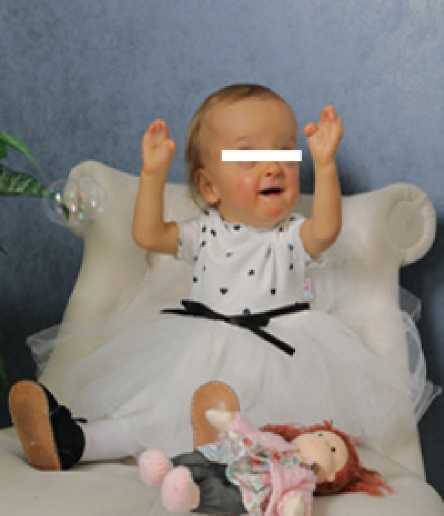
Особенности фенотипа пациентки Х.: «кукольное» лицо, низкорослость, синдактилия верхних конечностей
По результатам лабораторной диагностики патологии не выявлено. При выполнении рентгенограммы кистей выявлена аномалия развития обеих кистей, врожденная симбрахидактилия кистей и стоп, состояние после оперативного лечения (устранение синдактилии 1–2-го пальцев, отведение и противопоставление 1-го пальца правой кисти). При нейро-сонографии: легкое повышение периветрикулярной эхоплотности в лобно-теменно-затылочных областях без данных, свидетельствующих о врожденных пороках развития центральной нервной системы. Рыхлые полнокровные сплетения. Выраженная ассиметрич-ная бивентрикулодилатация за счет затылочных и височных рогов боковых желудочков (нейросоногра-фический контроль – без отрицательной динамики в сравнении с предыдущим исследованием). Муль-тиспиральная компьютерная томография головного мозга – субарахноидальное кровоизлияние по ходу намета мозжечка и задних отделах верхнего сагиттального синуса, внутрижелудочковое кровоизлияние I степени. Признаки синостоза латеральных отделов венечного шва с 2 сторон и лямбдовидного шва слева. Подозрение на синус-тромбоз. Для исключения тромбоза выполнена мультиспиральная компьютерная томография – ангиография, по результатам которой патологии не обнаружено. По данным комплексного ультразвукового исследования органов брюшной полости патологии не выявлено. Результаты кариотипирования показали нормальный женский кариотип (XX).
Пациентка Х. была консультирована специалистами. Заключение офтальмолога: «Задержка развития зрительных функций (центрального генеза). Содружественное альтернирующее расходящееся косоглазие с непостоянным углом девиации. OU-стеноз носослёзного канала». Заключение травматолога-ортопеда: «Врожденный подвывих головки лучевой кости справа. Дисплазия тазобедренных суставов». Заключение невролога: «Мышечная гипотония, контрактуры в тазобедренных суставах». Учитывая низкий иммунный статус ребенка, была проведена консультация врача-фтизиатра, поскольку ребенок входит в группу риска заболевания туберкулезом и подвержен значительному риску инфицирования микобактериями туберкулеза [7]. По результатам проведенного обследования данных, подтверждающих туберкулез органов дыхания, не получено. Для исключения локальной формы туберкулеза рекомендован скрининг 2 раза в год (до 8-летнего возраста постановка пробы Манту с 2 ТЕ, с 8 до 17 лет включительно – проба с аллергеном туберкулезным рекомбинантным, с 15 лет – диагностическая флюорография).
Обсуждение. Тяжесть синдрома Аперта может быть оценена на основе деформаций лицевого черепа, которые могут привести к осложнениям со стороны сердечно-сосудистой, мочеполовой и центральной нервной систем. Другими синдромами, связанными с синдактилией, являются следующие: синдром Корнелии де Ланге (микро-, брахицефалия, микрогения), Рассела – Сильвера (низкий рост, синдактилия), Смита – Лемли – Опица (микроцефалия, умственная отсталость, пороки сердца, легких, почек, пищеварительной системы, половых органов), Холт – Орама (синдром фокомелии, дисплазия соединительной ткани, аномалии верхних конечностей), Норман –
Робертса (гипоплазия мозжечка, гиппокампа, ствола мозга, микроцефалия, гипертелоризм, лимфедема), Фрейзера (криптофтальм, пороки развития половых органов, почек), Тричера Коллинза (грубые дефекты лицевого черепа, колобома век, тугоухость) и синдром Нагера (нижнечелюстной дизостоз, аномалии развития верхних конечностей) [8]. Клиническая картина связана с дефектами скелета, но не имеют связи с наличием краниосиностоза, именно поэтому дифференциальная диагностика синдрома Аперта затруднена из-за перекреста клинических признаков с другими синдромами [9].
Заключение. Учитывая генетическую природу синдрома Аперта, следует провести более глубокие исследования, чтобы понять, как он может отразиться на будущих поколениях. Это поможет выявить потенциальные риски и предложить соответствующие меры поддержки для таких детей и их семей. Ранняя пренатальная диагностика, особенно тяжелых форм заболеваний, очень важна для консультирования родителей относительно плохого прогноза этих случаев с серьезными последствиями. Комплексный подход к изучению, в том числе репродуктивного аспекта детей с акроцефалосиндактилией I типа, является критически важным для улучшения их качества жизни и достижения оптимальных результатов в области здравоохранения.
Вклад авторов. Все авторы сделали эквивалентный вклад в подготовку публикации.
