Использование газофазной химии и масс-спектрометрии для структурных исследований цистеинсодержащих пептидов

Автор: Миргородская О.А., Зубарев Р.A., Новиков А.В., Савельева Н.В., Краснов Н.В., Колуме Д., Роепсторфф П.
Журнал: Научное приборостроение @nauchnoe-priborostroenie
Рубрика: 25 лет институту аналитического приборостроения РАН
Статья в выпуске: 1 т.13, 2003 года.
Бесплатный доступ
На примере инсулина и цистеинсодержащих синтетических пептидов продемонстрирована возможность использования газофазных реакций для идентификации остатков цистеина и определения местоположения внутри- и межцепочечных дисульфидных связей. В качестве летучих реагентов использованы метиламин, аммиак и пентафтормасляная кислота.
Короткий адрес: https://sciup.org/14264269
IDR: 14264269 | УДК: 577.112.088.5:
The use of vapor phase chemistry and mass spectrometry for structural investigations of cysteine-containing peptides
A possibility of using vapor phase reactions for identification of cysteine residues and determination of location of the internal and external disulfide bonds was demonstrated by using insulin and cysteine-containing synthetic peptides. Methylamine, ammonia and pentafluoropropionic acid were used as volatile reagents.
Текст научной статьи Использование газофазной химии и масс-спектрометрии для структурных исследований цистеинсодержащих пептидов
Цистеины (Cys) играют существенную роль в структурной организации белков, природных пептидов и их синтетических аналогов, обеспечивая их биологическую функцию. В значительной степени это обусловлено способностью Cys к образованию дисульфидных связей.
При наличии двух и более Cys в последовательности возникает вопрос, в какой форме находятся остатки этих аминокислот, и если обнаруживается дисульфидная связь, то возникает дополнительный вопрос о ее местонахождении. Традиционно для анализа цистеинсодержащих соединений используется ферментативный гидролиз в сочетании с последующими химическими превращениями и масс-спектрометрической детекцией [1]. Однако достаточно часто именно наличие дисульфидных связей делает эти соединения устойчивыми к ферментативному гидролизу. Кроме того, как гидролиз, так и последующая модификация протекают в условиях (рН 7–8), когда возможно либо дополнительное образование дисульфидных связей, либо их перенос [2]. При гидролизе пептидных связей и при модификации в процессе идентификации как самих Cys, так и дисульфидных связей реакции осуществляют в водных растворах с использованием различных буферов для поддержания рН и избытков реагентов. В связи с этим, как правило, перед масс-спектрометрическими измерениями необходимо проводить очистку либо с помощью ВЭЖХ, либо с помощью электрофореза, либо какими-либо другими способами.
Для устранения большинства перечисленных недостатков при изучении структуры цистеинсодержащих соединений представляется целесообразным использовать газофазную химию с исполь- зованием летучих реагентов. Для гидролиза пептидных связей может быть успешно использована пентафтормаслянная кислота, а для разрушения дисульфидных связей — летучие амины, такие как метиламин и аммиак. В последнем случае при обработке аминами будут протекать две последовательные реакции: в-элиминирования и последующего присоединения аминов.
ОБСУЖДЕНИЕ ПРЕДЛАГАЕМОГО РЕШЕНИЯ И ЭКСПЕРИМЕНТАЛЬНЫХ РЕЗУЛЬТАТОВ
Реакция в —элиминирования достаточно широко используется для изучения белков, модифицированных остатками фосфорной кислоты или карбогидратами по Thr и Ser [3–5]. Для проведения реакций в —элиминирования используется обработка ферментативных гидролизатов белков в сильно шелочных водных средах, содержащих примерно 0.5 М NaOH [3, 4]. В результате этой реакции остатки карбогидратов и фосфорной кислоты удаляются, а модифицированные ранее ими остатки аминокислот Thr и Ser теряют воду. Образующиеся в результате реакции в -элиминирования продукты не являются стабильными соединениями, поскольку легко вступают в реакции со всевозможными аминами, присутствующими, как правило, в анализируемых смесях, что может затруднить интерпретацию даже при использовании масс-спектрометрии. Кроме того, сильнощелочная водная среда может приводить к частичному гидролизу пептидных связей. Очевидно также, что сильно солевая среда не является подходящей для последующего масс-спектрометрического анализа. В связи с этим должна быть введена дополнительная стадия обессоливания перед анализом.
Предложенные в работе [5] альтернативные ус- ловия проведения реакции β-элиминирования в газовой фазе для структурных исследований гликозилированных по Thr и Ser пептидов в газовой фазе в присутствии метиламина (NH2СН3) или аммиака (NH3) устраняют практически все недостатки, сопутствующие этой реакции в растворах. Преж- де всего в этом случае реакция протекает с образованием стабильных соединений. Анализируемые пробы не требуют дополнительной очистки.
Таким образом, реакцию взаимодействия цистинов с аминами можно описать приведенной ниже схемой:
CH 3
NH
CH 2
S
S
CH 2
NH C CO
H
NH C CO
NH 2 CH 3
H
NH 3 /NH 2 CH 3
CH 2
100Да
NH C CO
остаток цистина
стадия
(I)
стадия
(II)
NH 2
остаток
Dha, 69Да
NH 3
CH 2
NH C CO
H
86Да
Следует отметить, что на стадии I (непосредственно реакция β -элиминирования), как и в случае модифицированных (например, гликозилированных) по остатку Ser белков, образуется остаток дигидроаланина (Dha) [5]. В результате дальнейшей обработки на стадии II (addition reaction, реакция присоединения) метиламином (NH 2 СН 3 ) или аммиаком (NH 3 ) получаются продукты, молекулярные массы которых оказываются ниже в расчете на каждый аминокислотный остаток цистеина на 3 Дa или 17 Дa соответственно.
Также отметим, что привлекательным для использования такого подхода представляется тот факт, что анализируемый образец не требует последующей очистки. При этом для удаления следов аминов достаточно выдержать образцы в вакууме в течение 5 мин и затем растворить в подходящем для последующего масс-спектро-мерического анализа растворителе, как правило, содержащем либо ТFA, либо муравьиную кислоту. В результате модификации пептид дополнительно приобретает группы, которые способствуют присоединению протона, что должно увеличивать выход соответствующих ионов при масс-спектрометрической детекции (MALDI—MS, ESI—MS).
Возможность подобной обработки для удаления дисульфидных связей продемонстрирована в данной работе прежде всего на примере обработки метиламином инсулина человека.
В инсулине присутствуют 3 дисульфидных связи: одна внутрицепочечная в А-цепи между Cys6
и Cys11 и две межцепочечные связи между Cys7 А-цепи и Cys11 В-цепи, а также между Cys20 А-цепи и Cys19 В-цепи:
GIVEQCCTSICSLYQLENYCN
FVNQHLCGSHLVEALYLVCGERGFF YTPKT
Из результатов масс-спектрометрического анализа (рис. 1, а) видно, что все дисульфидные связи разрываются в достаточно мягких условиях при выдерживании при 56 оС в течение 30 мин, поскольку в спектре отсутствует исходный пептид и появляются продукты, соответствующие полной модификации каждой из цепей, т. е. А- и В-цепям с ожидаемыми значениями m / z = 2371.2 и m / z = = 3422.8 соответственно. Вместе с тем наличие иона с m / z = 2340.2 соответствует неполной модификации А-цепи инсулина, а именно превращению одного из остатков Cys, который в нативной молекуле скорее всего связан внутримолекулярной дисульфидной связью (Cys6 или Cys11), только в остаток Dha. С увеличением температуры до 70 оС этот промежуточный продукт исчезает, и модификация за то же самое время становится полной (рис. 1, б). Дальнейшее увеличение времени обработки при 70 оС до 1.5 ч приводит к частичному разрыву по пептидной связи Gly8—Ser9 в полностью модифицированной В-цепи (рис. 1, в). В масс-спектрах регистрируются соответственно пептид (9-30)-В-цепь с m / z = 2527.3 и метилированная (увеличенная масса на 14 Да) форма пеп-
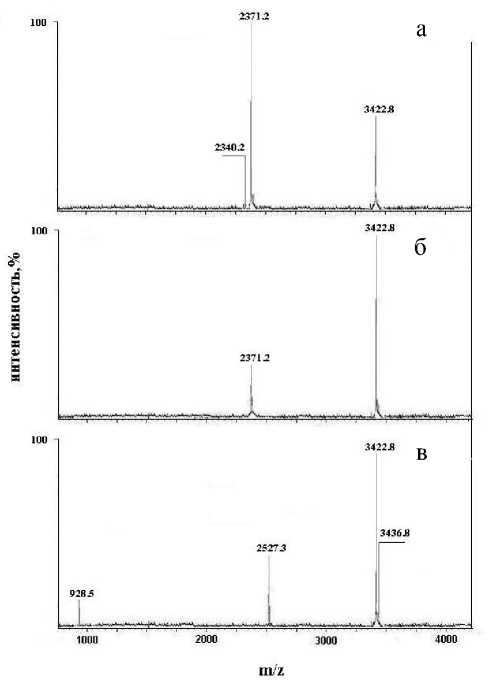
Рис. 1. MALDI-масс-спектры инсулина человека, подвергнутого газофазной обработке метиламином в условиях: а — 56 оС, 30 мин; б — 70 оС, 30 мин; в — 70 оС, 90 мин тида (1-8)-В-цепь (m/z = 928.5). В этих же условиях наблюдается также частичное метилирование всей полностью модифицированной В-цепи (m/z = = 3436.8).
После обработки аммиаком инсулина свиного (отличающегося от инсулина человека только заменой последнего аминокислотного остатка Thr в В-цепи на Ala) в жестких условиях, аналогичных приведенным на рис.1, б, наблюдается лишь частичная его модификация (таблица). Из представленных данных следует, что в реакционной смеси присутствуют как полностью модифицированные формы А- и В-цепей ( n = 1 и 4), так и их промежуточные формы ( n = 2, 3 и 5), в которых остатки Cys представлены в виде продуктов стадий I или II (см. схему реакции). Кроме того, регистрируется исходный инсулин ( n = 6), причем без каких-либо изменений. Из этого следует, что лимитирующей стадией процесса является разрыв обеих межмолекулярных дисульфидных связей.
Отметим, что существенно более интенсивные спектры относятся к А- и В-цепям, все цистеино- вые остатки (4 и 2 соответственно) которых оказываются модифицированными (n = 1 и 4).
При увеличении времени обработки аммиаком до 3 ч нативного инсулина зарегистрировано не было. Основными продуктами реакции являются полностью модифицированные А- и В-цепи при следовых количествах промежуточного продукта А-цепи с одной неприсоединенной NH 2 -группой ( n = 2).
Полученные данные указывают на то, что как реакция β-элиминирования, так и присоединения оказываются менее эффективными в случае использования аммиака для восстановления дисульфидных связей. Подобный результат демонстрировали эти реагенты в отношении дегликозилирования гликопептидов [5].
На основании того, что при обработке обоими реагентами инсулина обнаружены продукты неполной модификации А-цепи, в то время как В-цепь оказывается полностью модифицированной, можно предположить, что более стабильной оказывается внутримолекулярная дисульфидная связь в молекуле пептида.
Это предположение согласуется с данными эксперимента по обработке аммиаком природного пептида протегрина-1, содержащего две внутренние дисульфидные связи. Структура этого пептида: где * — амид, масса М mo = 2154.1 Да.

Как оказалось, в условиях (70 оС, 5 ч), когда в инсулине модифицируются все Cys, даже частичной модификации Cys в протегрине обнаружено не было.
Далее на примере двух синтетических пептидов с одной внутрицепочечной дисульфидной связью были изучены некоторые особенности их модификации аминами в газовой фазе. Для этого прежде всего был синтезирован аналог протегрина — укороченный пептид:
NPSCRRRFCK*(Р1), где * — амид, Мmo = 1264.6 Да.
Этот пептид был окислен с образованием одной дисульфидной связи (см. Экспериментальную часть). В результате окисления исходная масса пептида уменьшилась на 2 Да (рис. 2, а). Как показали результаты масс-спектрометрического анализа, модификация окисленного Р 1 метиламином и аммиаком при 70 оС протекает полностью в течение 30 мин и 1.5 ч соответственно (рис. 2, б, в). Исходная масса окисленного пептида соответственно уменьшается на 4 Дa при обработке метиламином и на 32 Дa аммиаком, что соответствует модификации обоих остатков, связанных дисульфидной связью.
Масс-спектрометрический анализ (ESI—MS) продуктов обработки инсулина свиньи аммиаком
|
Масс-спектрометричеcкие данные |
М, Дa* |
Продукты реакции (число модифицированных Cys в цепи) |
||||||
|
n |
m/z * |
I , % |
z |
Эксперим. |
Расчет |
Цепь |
I |
II |
|
1 |
1159.1 |
7 |
2 |
2316.2 |
2315.5 |
А |
— |
4 |
|
773.0 |
60 |
3 |
2316.0 |
|||||
|
579.9 |
18 |
4 |
2315.6 |
|||||
|
2 |
1150.1 |
7 |
2 |
2298.2 |
2298.5 |
А |
1 |
3 |
|
767.4 |
32 |
3 |
2299.2 |
|||||
|
575.7 |
7 |
4 |
2298.8 |
|||||
|
3 |
760.6 |
4 |
3 |
2278.8 |
2281.5 |
А |
2 |
2 |
|
571.5 |
23 |
4 |
2282.0 |
|||||
|
4 |
1122.6 |
10 |
3 |
3364.8 |
3365.8 |
В |
— |
2 |
|
842.6 |
77 |
4 |
3366.4 |
|||||
|
674.4 |
100 |
5 |
3367.0 |
|||||
|
562.0 |
68 |
6 |
3366.0 |
|||||
|
5 |
1117.5 |
5 |
3 |
3349.5 |
3348.8 |
В |
1 |
1 |
|
838.4 |
26 |
4 |
3349.6 |
|||||
|
670.8 |
34 |
5 |
3349.0 |
|||||
|
559.2 |
20 |
6 |
3349.2 |
|||||
|
6 |
1445.7 |
7 |
4 |
5778.8 |
5777.6 |
Исходный инсулин |
||
|
1156.7 |
24 |
5 |
5778.5 |
|||||
|
964.2 |
6 |
6 |
5779.2 |
|||||
*Приведены значения среднеизотопных масс.
чение m / z = 1650.7 для такого пептида совпадает с экспериментально определенным значением (рис. 3, г).
При реакции с йодацетамидом неокисленного пептида мы видим, что наряду с продуктом присоединения трех остатков ( m / z = 1766.7) присутствует заметное количество продукта, характеризующегося присоединением одного остатка йодацетамида и образованием одной дисульфидной связи ( m / z = 1650.7). В последнем случае, возможно, это обусловлено тем, что в условиях модификации при рН 7.8 трудно избежать частичного окисления, которое также легко осуществляется при этих же значениях рН.
В принципе при окислении пептида Р 2 в зависимости от местоположения дисульфидной связи возможно образование трех типов продуктов: Р 2 (Cys4–Cys9), Р 2 (Cys4–Cys13) или Р 2 (Cys9–Cys13). Поэтому прежде всего нужно определить, какой из них реально образуется при окислении. Для этого была использована обработка окисленного пептида в газовой фазе, но уже парами пентафтормасляной кислоты. Ранее в работах [6, 7] подобная обработка уже была использована для С-концевого

Рис. 2. MALDI-масс-спектры окисленного синтетического пептида Р1 до (а) и после газофазной обработки метиламином (б) и аммиаком (в) при 70 оС в продолжение 30 мин и 90 мин соответственно секвенирования пептидов, т.е. для образования серии продуктов, укороченных с С-конца. Хотя С-концевая аминокислота в пептиде Р2 присутствует изначально в виде амида, можно ожидать, что произойдет замена амидной на карбоксильную группу. Кроме того, можно было ожидать, что в процессе обработки под действием паров кислоты Asn также может превратиться в Asp. Тогда наряду с возможным образованием серии С-концевых продуктов следует ожидать разрыва связи Asp—Pro.
В реальных спектрах, полученных при гидролизе окисленного пептида Р 2 (рис. 4), мы видим, что действительно сам исходный пептид дезамидируется и масса окисленного пептида увеличивается на 2 Да ( m / z = 1595.7). Основным продуктом в гидролизате является дезамидированный пептид (1-10)-Р 2 (m/z = 1180.7), также присутствуют пептиды (1-9)-Р 2 ( m / z = 1065.7) и (3-10)-Р 2 ( m / z = = 1010.7). Кроме того, присутствуют в незначительном количестве дезамидированные пептиды (2-14)-Р 2 ( m / z = 1481.7) и (3-14)-Р 2 ( m / z = 1425.7). Из полученных данных следует, что как и ожидалось,
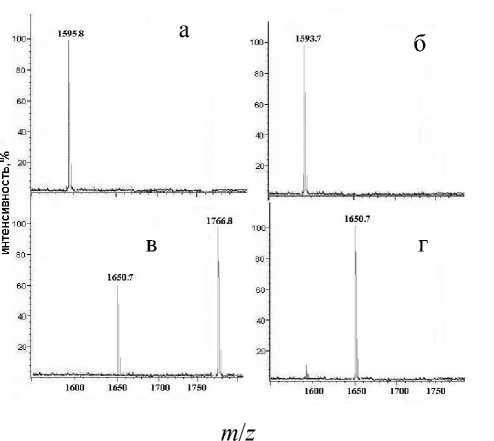
Рис. 3. MALDI-масс-спектры исходного (а) и окисленного (б) синтетических пептидов Р 2 до (а, б) и после (в, г) обработки йодацетамидом при 70 оС в продолжение 30 мин и 90 мин соответственно
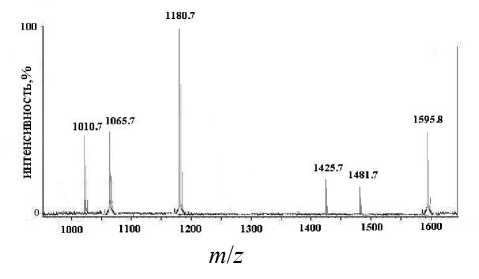
Рис. 4. MALDI-масс-спектр кислотного (PFPA, 70 оС, 3 ч) гидролизата синтетического пептида Р 2
при гидролизе происходит дезамидирование в исходном пептиде с последующим расщеплением связи Аsp—Pro и далее с отщеплением Asp как С-концевой аминокислоты. Кроме того, наблюдается гидролиз пептидных связей вблизи Gly.
Полученные данные четко указывают, что основным продуктом окисления является пептид Р 2 (Cys4—Cys9), поскольку основными продуктами гидролиза являются пептиды, содержащие Cys4— Cys9 дисульфидную связь. Это подтверждается также и отсутствием продуктов с массами, на 18 Да большими масс исходного пептида и пептидов (2-14)-Р 2 ( m / z = 1481.7 +18) и (3-14)-Р 2 ( m / z =
= 1425.7 + 18), что соответствовало бы расщеплению в них внутренней легко гидролизуемой пептидной связи Аsp—Pro.
Рассмотрим результаты обработки метиламином Р2 исходного, окисленного и обеих этих форм после соответствующей обработки йодацетамидом, т. е. всех форм пептида, масс-спектры которых представлены на рис. 3 а–г. После обработки метиламином при 70 оС в течение 30 мин по данным масс-спектрометрии оказалось, что во всех формах, включая исходную и полностью йодаце-тилированную, все 3 Cys подвергаются β-эли-минированию с последующим присоединением по двойной связи метиламина (рис. 3, д), давая основной ион с m / z = 1586.5. Наряду с этим ионом наблюдается ион с m / z = 1189.7 (GCRRRFCNPS), который соответствует (3-12)-Р 2 , в котором также все 3 Cys модифицированы.
Отметим, что в обоих пептидах Р1 и Р2, как и в случае инсулина, разрывы не происходят внутри пептидных фрагментов, в которых замкнута дисульфидная связь.
Таким образом, на основании полученных данных отметим, что обработка в газовой фазе аминами наиболее перспективна для обнаружения межцепочечных дисульфидных связей, но во всех случаях она также позволяет идентифицировать все формы Cys, присутствующие в пептидных цепях. Для идентификации местоположения дисульфидных связей успешно может быть использована обработка пептидов в газовой фазе пентафтормасляной кислотой. В этом случае можно идентифицировать и внутримолекулярные дисульфидные связи. Предложенные подходы позволяют проводить реакции в газовой фазе в одном реакторе для многих образцов, что делает эти процессы технологичными и пригодными для автоматизации, поскольку далее образцы без дополнительной очистки могут быть использованы для любого из масс-спектрометрических анализов: ESI—MS или MALDI—MS.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
В работе использовали инсулин человека и свиньи ("Sigma", США), протегрин-1 (PG-1), любезно предоставленный профессором В.Н. Кок-ряковым, метиламин (NH 2 CH 3 ), аммиак (NH 3 ), пентафтормасляную кислоту (PFPA), дитиотреит (DTT), иодацетамид (JAcNH 2 ), трифторуксусную кислоту (ТFA), уксусную кислоту, муравьиную кислоту и ацетонитрил (MeCN) производства фирмы "Merck", 2,5-дигидроксибензойную кислоту (DHB) и α-циано-4-гидроксикоричную кислоту (НССА) производства фирм "Serva" и "Aldrich" соответственно.
Синтез пептидов. Его осуществляли на твердофазном синтезаторе ("Intavis AG", Германия) аналогично схеме, описанной в работе [8]. Все пептиды были синтезированы в виде амидов. По завершении синтеза смолу промывали три раза дихлорметаном ("Merck", Германия) и высушивали в токе азота в течение 15 мин. Последующее отщепление пептида от смолы и снятие защитных боковых групп выполняли при 21оС в смеси TFA ("Fluka", Германия), триизопропилсилана и воды MilliQ ("Millipore", США) в объемном соотношении 95:2.5:2.5. По истечении двух часов пептиды осаждали холодным (0 оС) трет -бутилметиловым эфиром ("Fluka"). Пептидный преципитат перерас-творяли в 1 %-й водной уксусной кислоте и лиофилизовали.
Окисление пептидов. Осуществляли в атмосфере воздуха при встряхивании в 0.05 М карбонате аммония, рН 7.8, содержащем 10 % диметилсульфоксид. Концентрация пептидов при окислении составляла 1 мг/мл.
Восстановление дисульфидных связей и β-элиминирование пептидов. Восстановление дисульфидных связей дитиотреитом с последующим иодацетилированием осуществляли, как описано в работе [9].
Реакцию β-элиминирования пептидов осуществляли из твердого состояния в парах метиламина и аммиака. Предварительно анализируемые пептиды высушивали на вакуумной центрифуге в пластиковых пробирках вместимостью 0.5 мл (фирмы "Eppendorf"). Содержание пептида в каждой пробирке варьировали в пределах 1–5 нмоль для последующего исследования с помощью ESIMS и 20–50 пмоль — для MALDI—MS.
Высушенные в пробирках пептидные образцы помещали в реакционную камеру, представляющую собой стеклянный флакон вместимостью 8 или 22 мл, снабженный клапаном из инертного материала. В такой флакон ставили по 1 или 3–4 пробирки соответственно. Крышки с пробирок удаляли. Затем аккуратно по стенке вливали 150– 200 мкл 40 %-го водного метиламина или 25 %-го водного аммиака. Камеру заполняли аргоном, флакон закрывали и образцы инкубировали при температуре 56 или 70 оС в течение 0.5–5 ч. Следы растворителя удаляли центрифугированием на вакуумной центрифуге (5 мин).
Кислотный гидролиз. Для выполнения кислотного гидролиза высушенные в пробирках пептидные образцы помещали в реакционную камеру (см. выше), содержащую 100 мкл 20 %-й водной пентафтормасляной кислоты. Камеру заполняли аргоном и затем создавали вакуум до 1 мбар. Образцы гидролизовали в течение 60–90 мин при
-
90 оС. Следы растворителя удаляли центрифугированием на вакуумной центрифуге (10–15 мин).
Обработка инсулина свиного и протегрина-1 аммиаком. Инсулин свиной и протегрин-1 обрабатывали аммиаком, как описано выше, в реакционном флаконе вместимостью 8 мл при температуре 70 оС. В каждый флакон, содержащий 1 пробирку с образцом, добавляли 200 мкл 40 % водного метиламина или 25 %-го водного аммиака. Содержание протегрина-1 в каждой пробирке во всех опытах составляло 1 нмоль, время инкубации изменяли в пределах 0.5–5 ч. Обработку инсулина свиного осуществляли в продолжение 30 мин, при этом содержание пептида составляло 1, 2 и 5 нмоль в пробирке. Кроме того, для образца инсулина с содержанием 1 нмоль время обработки было увеличено до 3 ч.
ESI—MS анализ. Анализ проводили на приборе Finnigan MAT95 ("TermoQuest", Бремен, Германия), оборудованном электрораспылительным источником ионизации (electrospray ionization, ESI) и масс-анализатором с двойной фокусировкой. Все спектры получены в режиме съемки положительных ионов суммированием 100–150 импульсов. Объем анализируемой пробы 20 мкл. Скорость подачи раствора образца 1 мкл/мин.
Нативные и модифицированные пептиды растворяли в 50 %-м ацетонитриле в присутствии 0.1 %-й уксусной кислоты в концентрации 10– 20 пмоль/мкл.
MALDI—MS анализ. Для анализа этого типа использовался времяпролетный масс-спектрометр Applied Biosystems Voyager System 4270 ("Persep-tive Biosystems Inc.", Framingam, MA, США) с источником ионов замедленной экстракции в режиме отражения. В качестве матрицы использовали 2,5-дигидроксибензойную кислоту (25 г/л) и α-циано-4-гидроксикоричную кислоту (20 г/л) в смеси с ацетонитрилом, содержащим 0.1 %-ю трифторуксусную кислоту в объемном соотношении 70:30. Образцы приготовляли методом высушивания капли. Трифторуксусную кислоту (2 %, 0.3 мкл) смешивали с 0.3 мкл образца (0.5–2 пмоль на 1 мишень) и с 0.3 мкл раствора матрицы. В тексте и на рисунках приведены значения моноизо-топных масс, если не оговорено особо.
Список литературы Использование газофазной химии и масс-спектрометрии для структурных исследований цистеинсодержащих пептидов
- Фонтана А., Гросс Э. Практическая химия белка/Под ред. А. Дарбре. М.: Мир, 1989. 621 c.
- Торчинский Ю.М. Сульфидные и дисульфидные группы белков. М.: Наука, 1971. 229 с.
- 3. Oda Y., Nagasu T., Chait B.T. // Enrichment analysis of phosphorylated proteins as a tool for probing the phosphoproteome // Nat. Biotechnol. 2001. V. 19. P. 379-382.
- Meyer H.E., Hoffmann-Posorske E., Heilmeyer L.M. Jr. Determination and location of phosphoserine in proteins and peptides by conversion to S-ethylcysteine//Methods Enzymol. 1991. V. 201. P. 169-185.
- Mirgorodskaya E., Hassan H., Wandall H.H., Clausen H., Roepstorff P. Partial vapor-phase hydrolysis of peptide bonds: A method for mass spectrometric determination of O-glycosylated sites in glycopeptides//Anal. Biochem. 1999. V. 269. P. 54-65.
- Tsugita A., Takamoto K., Kamo M., Iwadate H. C-terminal sequencing of protein. A novel partial acid hydrolysis and analysis by mass spectrometry//Eur. J. Biochem. 1992. V. 206. P. 691-696.
- Mirgorodskaya O.A., Shevchenko A.A., Kamal Omer M. et al. Primary structure of three cationic peptides from porcine neutrophils//FEBS Lett. 1993. V. 330, N 3. P. 339-342.
- Kaselmann K.F., Budnik B.A., Kjeldsen F., Nielsen M.L., Olsen J.V., Zubarev R.A. Electronic ecitation gives informative fragmentation of polypeptide cations and anions//Eur. J. Mass. Spectrom. 2002. V. 8. P. 117-121.
- A practical guide to protein and peptide purification for microsequencing/Ed. Matsudaira P.T. Academic Press, San Diego, CA, 1989. 131 p.
