Исследование фармакокинетики, фармакодинамики и безопасности биоаналога пембролизумаба RPH-075 в сравнении с препаратом Китруда® у пациентов со злокачественными новообразованиями

Автор: Самойленко И.В., Покатаев И.А., Жукова Л.Г., Строяковский Д.Л., Орлова Р.В., Мудунов А.М., Пак М.Б., Зернова Е.В., Соболев А.В., Мочалова А.С., Алексеев Б.Я., Секачева М.И., Ледин Е.В., Петкова А.В., Ханонина Е.К., Подолякина А.И., Разживина В.А.
Журнал: Злокачественные опухоли @malignanttumors
Рубрика: Оригинальные исследования. Отечественные препараты
Статья в выпуске: 1 т.14, 2024 года.
Бесплатный доступ
Введение: Пембролизумаб представляет собой гуманизированное моноклональное антитело, селективно блокирующее взаимодействие между рецептором PD-1 и его лигандами. Препарат RPH-075 является биоаналогом оригинального препарата Китруда®.Цель: Целью данного исследования было установление эквивалентности фармакокинетических (ФК) свойств, а также показателей фармакодинамики (ФД), безопасности и иммуногенности препарата RPH-075 в сравнении с препаратом Китруда® у пациентов со злокачественными новообразованиями.Материалы иметоды: В настоящее многоцентровое двойное слепое рандомизированное исследование было включено 90 пациентов с меланомой и немелкоклеточным раком легкого, которые были рандомизированы в 2 группы терапии (RPH-075 и Китруда®) в соотношении 1:1. В обеих группах пембролизумаб применялся в виде монотерапии в дозе 200 мг внутривенно раз в 3 недели до наступления прогрессирования или непереносимой токсичности. Основной задачей в исследовании была оценка ФК после первого введения. Первичной конечной точкой оценки фармакокинетики был параметр AUC(0-504), безопасности - частота нежелательных реакций (НР). Решение об эквивалентности ФК свойств планировалось принять в случае, если границы двустороннего 90% доверительного интервала (ДИ) для отношения геометрических средних AUC(0-504) после однократного введения каждого из препаратов будут находиться в пределах 80,00-125,00%. Вторичными конечными точками были Сmax после первого введения, а также прочие параметры фармакокинетики, безопасности и иммуногенности. В рамках исследования также оценивались ФК и ФД параметры после многократного введения, и была запланирована пилотная оценка эффективности.Результаты: В статье приводится анализ данных первого этапа исследования (после первого введения препарата с периодом наблюдения в 3 недели). Анализ данных был заслепленным, поэтому группы были закодированы как А и Б; 90% ДИ для отношения средних геометрических AUC(0-504) после введения препарата А к AUC(0-504) препарата Б составил 93,50-121,16%, а для отношения Б к А - 82,54-106,95%. Полученные интервалы соответствуют заданному пределу эквивалентности 80,00-125,00%, на основании чего можно заключить, что препараты RPH-075 и Китруда® являются ФК эквивалентными. Оба препарата продемонстрировали сопоставимо высокое насыщение PD-1 рецепторов CD4+/CD8+ лимфоцитов по окончанию первого цикла (день 22). За анализируемый период связывающие антитела к пембролизумабу были выявлены у 2 пациентов (по одному в каждой группе), таким образом, оба препарата обладают сопоставимо низкой иммуногенностью. При анализе профиля безопасности за указанный период было зарегистрировано 7 НР у 4 пациентов в группе А и 4 НР у 3 пациентов в группе Б. Частота НР между группами статистически значимо не отличалась.Выводы: Показатели ФК, ФД, иммуногенности и безопасности биоаналога пембролизумаба RPH-075 были эквивалентны показателям оригинального препарата Китруда®.
Пембролизумаб, фармакокинетика, фармакодинамика, иммуногенность, безопасность, rph-075
Короткий адрес: https://sciup.org/140305809
IDR: 140305809 | DOI: 10.18027/2224-5057-2024-14-1-56-66
Study of the pharmacokinetics, pharmacodynamics, and safety of the biosimilar pembrolizumab RPH-075 compared to Keytruda® in patients with malignant neoplasms
Introduction: Pembrolizumab is a humanized monoclonal antibody selectively blocking the interaction between the PD-1 receptor and its ligands. The drug RPH-075 is a biosimilar to the original Keytruda®.Objective: To establish the equivalence of pharmacokinetic (PK) properties, as well as pharmacodynamic (PD) parameters, safety, and immunogenicity of the drug RPH-075 compared to Keytruda® in patients with malignant tumors.Materials and Methods: This multicenter double-blind randomized study included 90 patients with melanoma and non-small cell lung cancer who were randomized into two treatment groups (RPH-075 and Keytruda ®) in 1:1 ratio. In both groups, pembrolizumab was administered as monotherapy at a dose of 200 mg intravenously every 3 weeks until progression or intolerable toxicity. The primary aim of the study was to assess PK after the first administration. The primary endpoint for PK assessment was AUC(0-504), and for safety, it was the frequency of adverse events (AE). The decision on PK equivalence was planned to be made if the two-sided 90 % confidence interval (CI) for the geometric mean ratio of AUC(0-504) after a single administration of each drug would be within 80.00-125.00 %. Secondary endpoints included Cmax after the first administration, as well as the other PK, safety, and immunogenicity parameters. This study also assessed PK and PD parameters after multiple administrations, and a pilot efficacy assessment was planned.Results: This article presents the analysis of data from the first stage of the study (after the first drug administration with a 3-week observation period). The data analysis was blinded, and the treatment groups were coded as A and B. The 90 % CI for the geometric mean ratio of AUC(0-504) after the administration of drug A to AUC(0-504) of drug B was 93.50-121.16 %, and for the ratio of B to A, it was 82.54-106.95 %. The obtained intervals met the specified equivalence limit of 80.00-125.00 %, allowing us to conclude that RPH-075 and original Keytruda® are PK equivalent. Both drugs demonstrated comparably high saturation of PD-1 receptors on CD4+ / CD8+ lymphocytes at the end of the first cycle (day 22). Binding antibodies to pembrolizumab were detected in 2 patients (one in each group) over the analyzed period, indicating comparably low immunogenicity for both drugs. Safety profile analysis during this period revealed 7 AEs in 4 patients in group A and 4 AEs in 3 patients in group B. The frequency of AEs did not significantly differ between the groups.Conclusions: PK, PD, immunogenicity, and safety parameters of the pembrolizumab biosimilar RPH-075 were equivalent to those of the original Keytruda®.
Текст научной статьи Исследование фармакокинетики, фармакодинамики и безопасности биоаналога пембролизумаба RPH-075 в сравнении с препаратом Китруда® у пациентов со злокачественными новообразованиями
Ингибиторы контрольных точек иммунного ответа — одна из наиболее эффективных и перспективных групп среди иммуноонкологических препаратов. Ингибиторы контрольных точек отвечают за подавление иммунного ответа и сокращение числа активированных Т-лимфоцитов и их киллерной способности, например, для предотвращения избыточной стимуляции иммунитета и аутоиммунных реакций [1]. Рецептор PD-1 (programmed cell death 1) расположен на поверхности активированных Т-лимфоцитов и предотвращает активацию сигнального каскада PI3-киназы [2]. Это приводит к уменьшению пролиферации, цитотоксичности и высвобождения цитокинов из активированных Т-лимфоцитов [3]. В исследованиях взаимодействие между рецептором PD-1 на поверхности лимфоцитов и лигандом PD-L1 на поверхности опухолевых клеток блокировало иммунный ответ на эту опухоль, а блокада этого взаимодействия позволяло усилить реакцию Т-кле-ток и увеличить их противоопухолевую активность [4,5].
Пембролизумаб впервые в мире был зарегистрирован в 2014 году под торговым названием Китруда®. На момент подготовки настоящей статьи FDA (Федеральное Управление США по контролю за качеством пищевых продуктов и лекарственных средств) одобрило применение пембролизумаба в 39 показаниях в онкологии.
Биоаналог представляет собой биологический препарат, содержащий копию действующего вещества оригинального препарата [6,7], биоаналоги выделяются в отдельный класс веществ в связи со сложностью биологических молекул. Процесс производства биотехнологических препаратов (и препаратов на основе моноклональных антител в частности) многоэтапен и сложен, и особенности его организации могут привести к тому, что конечный продукт будет иметь отличия в профиле по сравнению с оригинальным препаратом [6,7], что в свою очередь может привести к формированию значимых различий в терапевтических свойствах препарата. В связи с этим, очень важным компонентом программы исследований биоаналогов является прямое сопоставление с оригинальным препаратом с целью доказательства аналогичности по параметрам качества, биологической активности, фармакокинетики, безопасности и эффективности [6,8,10,11]. Именно поэтому особенно важно в клинических исследованиях провести сравнительную оценку фармакокинетики, эффективности, безопасности и иммуногенности биоаналогичного препарата с оригинальным продуктом [12].
В месте с тем регистрация биоаналогов помогает повысить доступность терапии за счет снижения стоимости курса терапии [6,8] и обеспечения большего числа пациентов современной терапией [8,9].
RPH-075 представляет собой биоаналог препарата Китруда®, разработанный АО «Р-Фарм». Препарат RPH-075 по первичной аминокислотной последовательности, основным структурным характеристикам, профилю гликозилирования и параметрам специфической активности обладает высокой степенью сопоставимости с оригинальным препаратом Китруда®, он также продемонстрировал эквивалентность фармакокинетических и параметров безопасности в исследованиях in vivo на яванских макаках.
Основной целью настоящего клинического исследования было установление эквивалентности фармакокинетических свойств, а также сопоставимости показателей безопасности, иммуногенности и фармакодинамики препарата RPH-075 в сравнении с препаратом Китруда® после однократного (первого) внутривенного введения пациентам со злокачественными новообразованиями в качестве монотерапии 1 или 2 линии.
МАТЕРИАЛЫ И МЕТОДЫ
Дизайн
Настоящее исследование имело дизайн международного, многоцентрового двойного слепого рандомизированного исследования I фазы. Пациенты были рандомизированы в соотношении 1:1 в две группы терапии (группа препарата А и группа препарата Б). Буквенные обозначения для групп терапии были введены с целью сохранения за-слепления, так как к моменту проведения анализа данных исследование продолжается, в связи с этим разослепление групп терапии не проводилось.
Пациентам в обеих группах исследуемый препарат (RPH-075 или Китруда®) вводили в фиксированной дозе 200 мг в виде 30-минутной внутривенной инфузии один раз в 3 недели. Терапия продолжалась до наступления прогрессирования или непереносимой токсичности.
ПАЦИЕНТЫ
В исследование включали пациентов мужского и женского пола в возрасте старше 18 лет со следующими злокачественными новообразованиями: меланома кожи (впервые выявленная и не леченная ранее нерезектабельная или метастатическая, или при прогрессировании после проведенной ранее неоадъювантной/адъювантной терапии, или после проведенной ранее терапии 1 линии нерезектабельная или метастатическая меланома кожи), плоскоклеточный немелкоклеточный рак легкого (впервые выявленный нерезектабельный или метастатический с уровнем экспрессии PD-L1 ≥ 1 %, с непереносимостью химиотерапии 1 линии или с прогрессированием на фоне противоопухолевой терапии 1 линии) и нерезектабельный или метастатический плоскоклеточный рак головы и шеи с уровнем экспрессии PD-L1 TPS ≥ 50% с прогрессированием на фоне или после платиносодержащей химиотерапии 1 линии. Таким образом, пембролизумаб применялся в качестве терапии 1 или 2 линии. Основными критериями включения в исследование были также: масса тела от 50 до 100 кг, балл по шкале ECOG 0–2, ожидаемая продолжительность жизни не менее 12 недель, наличие измеримых контрольных опухолевых очагов согласно критериям RECIST 1.1, т. к. в исследовании также планировалась пилотная оценка эффективности. Не допускалось участие пациентов, ранее получавших терапию пембро-лизумабом и другими анти-PD-1/PD-L1/PD-L2 препаратами (предшествующая иммунотерапия в адъювантном режиме, кроме терапии пембролизумабом, не являлась критерием невключения), пациентов с увеальной меланомой или меланомой слизистых оболочек, с прогрессирующими или сопровождающимися клиническими симптомами метастазами в центральной нервной системе, с тяжелыми сопутствующими заболеваниями, с угрожающими жизни остро развивающимися осложнениями основного заболевания, с системными аутоиммунными заболеваниями в активной фазе, а также пациентов, получающих постоянно ГКС и другие препараты с иммуносупрессивным действием.
Этические аспекты
До проведения процедур в рамках исследования пациентам было предоставлено полное описание характера, цели, содержания, процедуры исследования и любых НР, которые могли возникнуть в ходе него. Все пациенты дали добровольное согласие на участие в исследовании и подписали форму информированного согласия до его начала.
Исследование было проведено в полном соответствии с протоколом исследования, требованиями ICH GCP E6 (R2), Правилами надлежащей клинической практики Евразийского экономического союза (Решение Совета ЕЭК № 79 от 03 ноября 2016 г.), этическими принципами Хельсинкской декларации последнего пересмотра, а также прочими действующими регуляторными требованиями Российской Федерации и Евразийского Экономического Союза.
ОЦЕНКАФАРМАКОКИНЕТИКИ
Для анализа ФК после первого введения биообразцы (по 6 мл крови) у пациентов отбирали по следующей схеме: непосредственно перед первой инфузией, сразу после окончания инфузии, а далее через 2 ч, 4 ч, 5 ч, 6 ч, 8 ч, 12 ч, 24 ч, 48 ч, 72 ч, 120 ч, 168 ч, 336 ч и 504 ч после окончания первой инфузии. В последней точке забор проводился перед вторым введением препарата. Из отобранных биообразцов получали сыворотку, которую замораживали и хранили при температуре не выше –65°С.
Для количественного определения пембролизумаба в образцах сыворотки крови применялась валидированная методика гетерофазного иммуноферментного анализа (ИФА) с использованием коммерческого набора KRISHGEN BioSystems KRIBIOLISA™ Pembrolizumab (KEYTRUDA®) ELISA. Нижний предел количественного определения методики составил 2 мкг/мл.
Первичной конечной точкой оценки ФК в рамках исследования была площадь под фармакокинетической кривой «концентрация-время» пембролизумаба после первого (однократного) введения, усеченная до второго введения, т. е. точки 504 ч (AUC (0–504) ). Вторичной конечной точкой после первого введения была максимальная сывороточная концентрация пембролизумаба после первого введения (С max ). Дополнительно оценивались также следующие ФК-параметры после первого введения: AUC (0–∞) , время достижения максимальной концентрации (T max ), период полувыведения пембро-лизумаба (T 1/2 ), объем распределения (V d ), константа скорости элиминации (K el ).
Оценка фармакодинамики
Отбор биообразцов (по 5 мл крови) для анализа ФД проводили непосредственно перед первой и второй инфузиями (менее чем за 30 минут до инфузии). Для анализа использовались образцы цельной крови.
Поисковой конечной точки для оценки ФД был уровень насыщенности пембролизумабом PD-1 рецепторов CD4+/CD8+ -лимфоцитов периферической крови. Детектирование насыщенности рецепторов проводили с помощью валидированной методики определения отношения доли Т-клеток, с которыми связались моноклональные антитела анти-PD-1, меченные флуоресцентным красителем фикоэритрином, после введения исследуемых препаратов к доле T-клеток, экспрессирующих PD-1, в образцах цельной периферической крови пациентов до введения препаратов.
Оценка безопасности
Для оценки безопасности проводились: регулярный сбор данных о жалобах и симптомах, физикальный осмотр, оценка витальных показателей (измерение температуры тела, артериального давления, пульса), контроль лабо- раторных показателей (клинический и биохимический анализы крови, коагулограмма, анализ крови на гормоны щитовидной железы, общий анализ мочи), электрокардиограмма (ЭКГ), оценка статуса по шкале ECOG. Нежелательные явления (НЯ) регистрировались с момента первого введения пембролизумаба. Тяжесть НЯ оценивалась на основе общепринятых терминологических критериев CTCAE 5.0.
Оценка иммуногенности
Отбор биообразцов (по 6 мл крови) для анализа иммуногенности проводили непосредственно перед первой и второй инфузиями. Из отобранных биообразцов получали сыворотку, которую замораживали и хранили при температуре не выше –65°С.
Для оценки иммуногенности была разработана и валидирована биоаналитическая методика полуколичествен-ного определения антител к пембролизумабу в сыворотке крови человека при помощи иммуноферментного анализа (ИФА) с применением коммерческого набора KRIBIOLISATM Anti-Pembrolizumab (KEYTRUDA®) ELISA.
Расчет размера выборки и статистическая методология
В соответствии с российскими и международными рекомендациями, расчет размера выборки для оценки биоэквивалентности фармакокинетических свойств исследуемых препаратов основан на коэффициенте межсубъектной вариации ФК параметра AUC. На основе литературных данных, значение таргетного коэффициента вариации было принято как равное 30% [13]. Для исследования был выбран параллельный дизайн, уровень ошибки 1 рода alfa был принят за 0,05 (односторонняя), мощность тестирования гипотезы биоэквивалентности составила 80%. Таким образом, было установлено, что размер выборки составляет не менее 76 пациентов. С учетом возможного выбывания пациентов в ходе исследования на уровне 10%, необходимо было рандомизировать не менее 84 пациентов.
Статистический анализ был проведен под руководством ответственного биостатистика, в соответствии с требованиями ICH, а также другими применимыми требованиями и законами. До проведения статистического анализа был утвержден План статистического анализа.
Анализ биоэквивалентности проводился исходя из предположения о лог-нормальном распределении параметров. Для вычисления 90% ДИ значения анализируемых показателей логарифмировались, затем проводился дисперсионный анализ ANOVA с использованием статистического пакета R.
Решение об эквивалентности ФК свойств планировалось принять в случае, если границы двустороннего 90% ДИ для отношения геометрических средних AUC (0 _ 504) после однократного введения каждого из препаратов будут находятся в пределах 80,00–125,00%.
РЕЗУЛЬТАТЫ
Демографические данные
В исследование было скринировано 116 пациентов, 26 человек не соответствовали критериям отбора в исследование, а 90 пациентов успешно прошли скрининг и были рандомизированы в 2 группы терапии: группа А (n = 47) и группа Б (n = 43). Одному пациенту при первом введении был введен препарат не в соответствии с группой рандомизации, в связи с чем его данные для оценки первичной конечной точки были учтены в группе фактически введенного препарата.
Медиана возраста в группе препарата А составила 65,0 лет (диапазон от 38,0 до 87,0 лет), а в группе Б — 67,0 лет (диапазон от 40,0 до 83,0 лет). Все рандомизированные пациенты были представителями европеоидной расы. В обеих группах терапии преобладали пациенты женского пола (52,08% в группе А и 69,05% в группе Б).
Таблица 1. Демографические данные пациентов (n = 90)
Table 1. Patients' demographics (n = 90)
|
Показатель |
Препарат А (n = 48) |
Препарат Б (n = 42) |
|
Пол |
||
|
Женский (%) |
25 (52,08) |
29 (69,05) |
|
Мужской (% |
23 (47,92) |
13 (30,95) |
|
Раса |
||
|
Европеоидная (%) |
48 (100,00) |
42 (100,00) |
|
Возраст (лет) * |
65,0 (38,0 ± 87,0) |
67,0 (40,0 ± 83,0) |
|
Рост (см) * |
167,56 (± 9,75) |
166,71 (± 10,75) |
|
Масса тела (кг) * |
75,54 (± 11,95) |
77,89 (± 10,49) |
|
Первоначальный диагноз |
||
|
Меланома (%) |
46 (95,83) |
42 (100,00) |
|
Немелкоклеточный рак легкого (%) |
2 (4,17) |
0 (0,00) |
|
Тип заболевания |
||
|
Метастатический |
12 (25,00) |
12 (28,57) |
|
Нерезектабельный |
36 (75,00) |
30 (71,43) |
|
Предшествующая терапия |
||
|
Адьювантная |
10 (20,83) |
11 (26,19) |
|
Неоадьювантная |
2 (4,17) |
1 (2,38) |
|
Лучевая |
1 (2,08) |
3 (7,14) |
|
Хирургическое лечение |
39 (81,25) |
38 (90,48) |
|
Системная терапия 1 линии |
9 (18,75) |
6 (14,29) |
|
Терапия ингибиторами BRAF/MEK |
3 (6,25) |
4 (9,52) |
|
Химиотерапия |
5 (10,42) |
1 (2,38) |
|
Другое |
1 (2,08) |
1 (2,38) |
В таблице приведены средние показатели (стандартное отклонение).
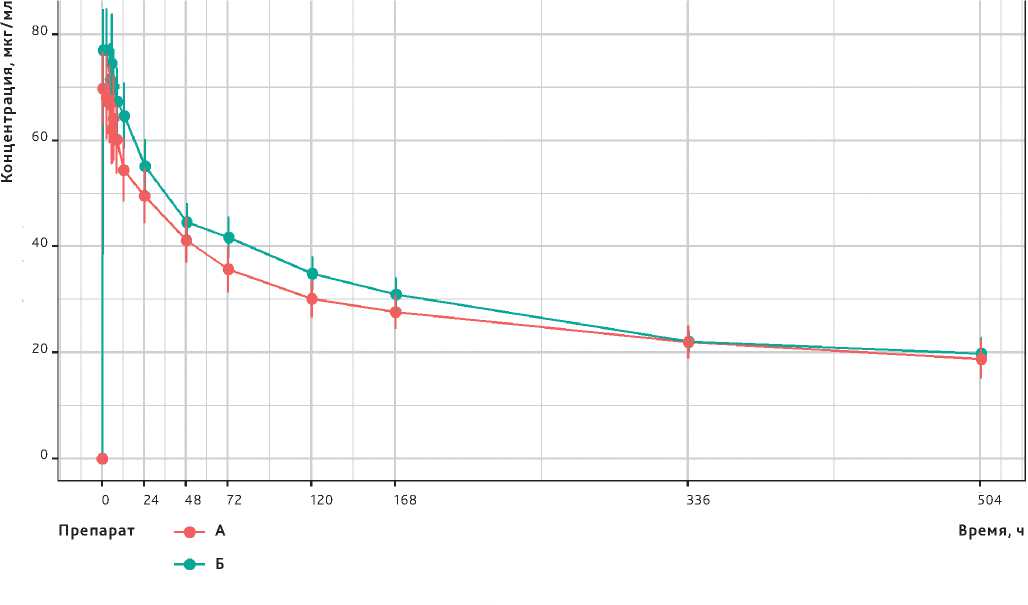
Рисунок 1. Динамика концентраций пембролизумаба (представлены среднее ± 95% ДИ) после первого введения каждого из исследуемых препаратов (n = 90)
Figure 1. Dynamics of pembrolizumab concentrations (mean ± 95 % CI presented) after the first administration of each drug (n = 90)
Обе группы были сопоставимы по антропометрическим, демографическим данным и исходным характеристикам заболевания (р > 0,05 для всех показателей показателей, табл. 1).
Почти все пациенты в группе препарата А (46/48, 95,83%) и все пациенты в группе препарата Б (42/42, 100,00%) имели диагноз «меланома», и только у 2 пациентов в группе А был диагностирован немелкоклеточный рак легкого.
Фармакокинетика
Анализ фармакокинетики проводился после первого введения пембролизумаба в течение периода в 22 дня (504 часа). В анализ ФК, согласно протоколу, были включены данные пациентов, получивших как минимум одно введение одного из исследуемых препаратов, у которых на протяжении первого цикла терапии (дни 1–22 исследования) было пропущено не менее 3 точек отбора биообразцов.
Таблица 2. Рассчитанные ключевые ФК-параметры пембролизумаба после первого введения каждого из исследуемых препаратов (n = 90)
Table 2. Key PK parameters of pembrolizumab calculated after the first administration of each drug (n = 90)
|
ФК параметры |
Препарат А (n = 48) |
Препарат Б (n = 42) |
Значение p1 |
|
AUC (0–504) , (мкг/мл) × ч |
14487,62 ± 4138,42 |
13916,07 ± 4701,65 |
0,513 |
|
C max , мкг/мл |
98,91 ± 32,83 |
86,27 ± 27,79 |
0,090 |
|
AUC (0–∞) , (мкг/мл) ×ч |
27607,92 ± 17094,41 |
26530,58 ± 12881,7 |
0,699 |
|
T max , ч |
8,88 ± 24,35 |
16,63 ± 77,42 |
0,330 |
|
T 1/2 , ч |
423,45 ± 297,84 |
460,33 ± 265,33 |
0,404 |
|
Cl, мл/ч |
15,36 ± 6,19 |
16,94 ± 9,2 |
0,513 |
|
K el , сут -1 |
0,05 ± 0,03 |
0,05 ± 0,02 |
0,404 |
|
V d , мл |
2870,15 ± 961,6 |
3474,58 ± 1838,86 |
0,067 |
Представлены средние ± стандартное отклонение.
-
1 Уровень значимости при сравнении показателей после препаратов (Т-критерий Вилкоксона).
Таблица 3. Результаты оценки ФК эквивалентности по первичной конечной точке AUC (0-504 ) (n = 90)
Table 3. Results of PK equivalence assessment for the primary endpoint AUC (0 50.) (n= 90)
|
Препараты |
Параметр |
CVinter1 |
T/R |
Рассчитанные 90 % ДИ |
Допустимые значения для 90% ДИ |
Биоэквивалентность |
|
А/Б |
AUC (0-504) |
37,98% |
1,0644 |
93,5–121,16% |
80,00–125,00% |
Подтверждена |
|
Б/А |
AUC (0-504) |
37,98% |
0,9395 |
82,54–106,95% |
80,00–125,00% |
Подтверждена |
1 CVinter — межиндивидуальный коэффициент вариации; ДИ — доверительные интервалы.
А / Б — проверка эквивалентности препарата А по отношению к препарату Б.
Б /А — проверка эквивалентности препарата Б по отношению к препарату А.
В данную популяцию вошли все 90 рандомизированных пациентов.
Динамика концентраций пембролизумаба в реальной временной шкале после введения исследуемых препаратов представлена на рисунке 1, а рассчитанные основные ФК-параметры — в таблице 2. Из представленных данных следует, что ФК показатели в группах, рассчитанные на основании концентраций пембролизумаба в сыворотке крови, после введения препаратов RPH-075 и Китруда® не имеют статистически значимых отличий.
После расчета ФК параметров было проведено статистическое определение ФК эквивалентности (биоэквивалентности) по первичной конечной точке с применением ANOVA, по результатам которого было установлено, что 90% ДИ для отношения средних геометрических AUC (0-504 ) препарата А к AUC (0-504 ) препарата Б составил 93,50-121,16%, а для отношения Б к А — 82,54–106,95%. Полученные интервалы соответствуют установленному пределу эквивалентности для AUC (0—504) — 80,00–125,00%, на основании чего можно заключить, что препараты RPH-075 и Китруда® обладают эквивалентной ФК при внутривенном введении.
Фармакодинамика
Медиана насыщенности пембролизумабом рецепторов PD-1 CD4+/CD8+ -лимфоцитов в обеих группах терапии составила более 90%. Медиана насыщенности пембро-лизумабом рецепторов PD-1 CD4+ -лимфоцитов в группе А составила 94,38% [90,00; 96,74], в группе Б 94,75% [90,86; 97,33]. Медиана насыщенности пембролизумабом рецепторов PD-1 CD8+ -лимфоцитов в группе А составила 93,48% [86,76; 96,83], в группе Б — 94,60% [86,16; 97,05]. При этом статистически значимой разницы между группами терапии не было выявлено как для CD4+ , так и для CD8+ лимфоцитов. Таким образом, эквивалентность параметров ФК препаратов RPH-075 и Китруда® также была подтверждена.
Безопасность
За анализируемый период (дни с 1 по 22 исследования) НР были выявлены примерно у половины пациентов в каждой группе терапии: у 20/48 (41,67%) пациентов в группе препарата А и у 21/42 (50,00%) в группе препарата Б. В группе препарата А были зарегистрированы 7 НР у 4/48
(8,33%) пациентов, в группе препарата Б — 4 НР у 3/42 (7,14%) пациентов. Частота НР между группами была сопоставимой. НР были представлены астенией, артралгией, анемией, повышением уровня аланинаминотрансферазы, гипертиреозом, эритематозной сыпью, пирексией, повышением уровня билирубина в крови.
Большинство НР были 1–2 степени тяжести. НР ≥ 3 степени тяжести были зафиксированы у 3/48 (6,25%) пациентов в группе препарата А и у 4/42 (9,52%) пациентов в группе препарата Б. НР 3 степени тяжести была зафиксирована только у одного пациента в группе препарата Б (повышение уровня билирубина в крови), остальные НР имели 1–2 степени тяжести. Иммуноопосредованная НР была зарегистрирована только у 1 пациента в группе препарата А и была представлена эритематозной сыпью 1 степени тяжести. Сводные данные по безопасности представлены в таблице 4.
Серьезные нежелательные явления (СНЯ) были зарегистрированы у 4 пациентов (у 2 в группе препарата А и у 2 в группе препарата Б), события не имели связи с исследуемой терапией. За анализируемый период было зафиксировано 2 летальных исхода, по одному в каждой группе терапии: легочная эмболия в группе А и желудочнокишечное кровотечение в группе Б, оба события не имели связи с исследуемой терапией.
Таким образом, можно заключить, что препараты RPH-075 и Китруда® обладают аналогичными характеристиками безопасности.
Иммуногенность
По результатам проведенного анализа определения иммуногенности связывающие антитела были обнаружены у 2 пациентов (по одному в каждой группе терапии). Нужно отметить, что у одного пациента были обнаружены связывающие антитела к пембролизумабу в первой точке забора (до введения препарата), при этом применения пембролизумаба до включения в исследование в анамнезе пациента не зафиксировано. Появление связывающих антител к пембролизумабу было расценено как связанное с кросс-реактивностью и иммунным ответом на предшествующую терапию. Нейтрализующей активности обнаруженных связывающих антител не было выявлено ни в одном из случаев.
Таблица 4. Резюме нежелательных явлений
Table 4. Adverse events
СНЯ — серьезное нежелательное явление; НР — нежелательная реакция; ННР — непредвиденная нежелательная реакция;
СНР — серьезная нежелательная реакция; СННР — серьезная непредвиденная нежелательная реакция; иоНР — иммуноопосредован-ная нежелательная реакция; СКФ — скорость клубочковой фильтрации; АСТ — аспартатаминотрансфераза.
ОБСУЖДЕНИЕ
RPH-075 представляет собой биоаналог оригинального иммуноонкологического препарата Китруда®, разработанный АО «Р-Фарм».
Концепция клинической разработки биоаналога предполагает пошаговое исследование ФК (и ФД) сопоставимости (т. е. проведение исследования I фазы/биоэквивалент-ность) с последующим исследованием сопоставимости эффективности и безопасности, включая оценку иммуногенности (т. е. III фазу). Это исследование представляет собой 1 этап клинической разработки биоаналога RPH-075, то есть сравнение ФК, ФД свойств, показателей безопасности и иммуногенности.
Методика проведения клинического исследования, а именно его дизайн, выбор конечных точек, методы оценки эффективности и безопасности, статистические методы, соответствует российским и международным стандартам (Правилам проведения исследований биологических лекарственных средств ЕАЭС, а также соответствующим международным рекомендациями EMA и FDA), а также максимально благоприятному профилю риск/польза для участников исследования. В соответствии с указанными рекомендациями был выбран дизайн рандомизированного, двойного слепого исследования в параллельных группах. Исследование проводилось в популяции пациентов с различными злокачественными новообразованиями, которым показана терапия пембро-лизумабом. Исследование на пациентах оправдано, так как применение пембролизумаба ассоциировано с серьезными иммуноопосредованными НР, в связи с чем применение данного препарата у здоровых добровольцев неэтично. Размер выборки определялся исходя из значе- ния коэффициента вариации ключевого ФК-показателя, что соответствует международным стандартам оценки биоэквивалентности.
Первичная конечная точка оценивалась после однократного (первого) введения исследуемых препаратов, в результате чего была подтверждена ФК эквивалентность (биоэквивалентность) препаратов RPH-075 и Китруда® на основании того, что рассчитанные 90% ДИ как для отношений AUC (0 _ 504) как препарата А/Б, так и Б/А (разослепле-ние данных не проводилось) уложились в установленные пределы эквивалентности 80,00–125,00%. Этот факт является ключевым моментом в оценке первого этапа — фармакокинетической эквивалентности биоаналога.
Анализ безопасности и иммуногенности проведен за относительно небольшой период времени. Однако по результатам оценки первого цикла терапии можно заключить, что все НР были ожидаемы и описаны в литературных данных о профиле безопасности пемброли-зумаба.
В настоящее время исследование продолжается, продолжается сбор долгосрочных данных ФК, ФД, безопасности и иммуногенности, а также запланирована пилотная оценка эффективности. В исследовании предусмотрено постоянное тщательное наблюдение за пациентами.
Коллектив авторов выражает особую благодарность коллегам, принимавшим неп осредственное участие в исследовании: К. Д. Пеньков, С. А. Проценко , А. И. Семенова, Е.А. Готовкин, О. Абдельгафур, Ю.Ю. Макарычева, А.А. Кельн, Т.Т. Андабеков, В.В. Чистякова, А.В. Султанбаев, Р. М. Давыдкин, А. В. Хоринко, Я. С. Чапко, А. А. Гофман, И.О. Белогорцев, В.К. Лядов, О.Б. Талибов, М.Г. Чернобровкин, А.А. Андрианов, Т.О. Попова, Р.Ю. Кобзев, М.Ю. Самсонов, О. В. Филон.
Список литературы Исследование фармакокинетики, фармакодинамики и безопасности биоаналога пембролизумаба RPH-075 в сравнении с препаратом Китруда® у пациентов со злокачественными новообразованиями
- Царев И.Л., Мелерзанов А.В. Обзор подходов к иммунотерапии в онкологии. Исследования и практика в медицине 2017;4(3):51-65. https://doi.org/10.17709/2409-2231-2017-4-3-5
- Parry R.V., Chemnitz J.M., Frauwirth K.A., et al. CTLA-4 and PD-1 receptors inhibit T-cell activation by distinct mechanisms. Mol Cell Biol 2005;25(21):9543-53. https://doi.org/10.1128/MCB.25.21.9543-9553.2005
- Keir M.E., Butte M.J., Freeman G.J., Sharpe A.H. PD-1 and its ligands in tolerance and immunity. Annu Rev Immunol 2008;26:677-704. https://doi.org/10.1146/annurev.immunol.26.021607.090331
- Mellman I., Coukos G., Dranoff G. Cancer immunotherapy comes of age. Nature 2011;480(7378):480-9. https://doi.org/10.1038/nature10673
- Hamanishi J., Mandai M., Konishi I. Immune checkpoint inhibition in ovarian cancer. Int Immunol 2016;28(7):339-48. https://doi.org/10.1093/intimm/dxw020
- Zhang H., Wu M., Zhu X., et al. A phase I, randomized, single-dose study to evaluate the biosimilarity of QL1206 to denosumab among Chinese healthy subjects. Front Pharmacol 2020;11:01329. https://doi.org/10.3389/fphar.2020.01329
- Zhang H., Li C., Liu J., et al. Safety and pharmacokinetics of a biosimilar of denosumab (KN012): Phase 1 and bioequivalence study in healthy Chinese subjects. Expert Opin Investig Drugs 2021;30(2):185-192. https://doi.org/10.1080/13543784.2021.1863371
- Farahani M.F., Maghzi P., Aryan N.J., et al. A randomized, double-blind, parallel pharmacokinetic study comparing the trastuzumab biosimilar candidate, AryoTrust®, and reference trastuzumab in healthy subjects. Expert Opin Investig Drugs 2020;29(12):1443-1450. https://doi.org/10.1080/13543784.2020.1831470
- Bushra R., Shoaib M.H., Ali H., Ghayas S. Pharmacokinetics and bioequivalence assessment of optimized directly compressible Aceclofenac (100 mg) tablet formulation in healthy human subjects. PLoS One 2020;15(9):e0238951. https://doi.org/10.1371/journal.pone.0238951
- Zhu X., Ding Y., Yu Y., et al. A Phase 1 randomized study compare the pharmacokinetics, safety and immunogenicity of HLX02 to reference CN- and EU-sourced trastuzumab in healthy subjects. Cancer Chemother Pharmacol 2021;87(3):349-359. https://doi.org/10.1007/s00280-020-04196-9
- Finck B., Tang H., Civoli F., et al. Pharmacokinetic and pharmacodynamic equivalence of pegfilgrastim-cbqv and pegfilgrastim in healthy subjects. Adv Ther. 2020;37(10):4291-4307. https://doi.org/10.1007/s12325-020-01459-y
- Shin D., Lee Y.J., Choi J., et al. A phase I, randomized, single-dose pharmacokinetic study comparing sb8 (bevacizumab biosimilar) with reference bevacizumab in healthy volunteers. Cancer Chemother Pharmacol 2020;86(4):567-575. https://doi.org/10.1007/s00280-020-04144-7
- Freshwater T., Kondic A., Ahamadi M., et al. Evaluation of dosing strategy for pembrolizumab for oncology indications. J Immunother Cancer 2017;5:43. https://doi.org/10.1186/s40425-017-0242-5
