Исследование равновесий в неводных растворах поликислот (модель и эксперимент)
")
Автор: Танганов Б.Б.
Журнал: Вестник Восточно-Сибирского государственного университета технологий и управления @vestnik-esstu
Статья в выпуске: 3 (34), 2011 года.
Бесплатный доступ
Разработана модель расчета термодинамических констант диссоциации четырехосновных кислот при совместной нейтрализации первой-второй и третьей-четвертой карбоксильных групп в растворителе. Обоснован метод Дэвиса при оценке коэффициентов активности всех ионов в титруемой системе.
Многоосновные кислоты, термодинамические константы диссоциации, логарифмические диаграммы
Короткий адрес: https://sciup.org/142142376
IDR: 142142376 | УДК: 544.363.081
Equilibrium in nonaqueous solutions of polyacid (model and experiment)
A model _ for calculating the thermodynamic dissociation constants chetyrehosnovnyh neutralize acids in the joint first-second-and third-fourth of the carboxyl groups in the solvent. Davis grounded method for assessing the activity coefficients of ions in the titrated system.
Текст научной статьи Исследование равновесий в неводных растворах поликислот (модель и эксперимент)
Многоосновные кислоты являются исходными соединениями в синтезе термостойких полимеров. Существующие методы расчета и определения констант диссоциации многоосновных кислот, предложенные Спикменом [1], позднее Бейтсом [2], справедливы для водных растворов. Определение ступенчатых констант диссоциации многоосновных кислот в неводных растворах представляет значительную трудность из-за невозможности прямого определения активности ионов водорода (для простоты рассуждений - концентрации ионов водорода [ H + ]) в каждой точке титрования. Поликислоты обычно характеризуются близостью констант диссоциации. Поэтому число скачков на кривых потенциометрического титрования не соответствует числу функциональных групп многоосновных кислот, что затрудняет непосредственное определение нескольких рК . Отметим, что для раздельного определения констант диссоциации поликислот (или смеси электролитов) необходимо, чтобы отношение ближайших констант диссоциации было не менее 1000.
Представим равновесия в растворах поликислот
H n A = H (n - 1) A - + H
H (n - i) A - = H (n — 2) A 2 + H + (1)
HA(1 - n) = An- + H+
Константы диссоциации последовательно запишем в виде:
K 1 = [H+] ⋅ ([H (n - 1) A] - /[H n A]) ⋅ f 1 /f 0
K2 = [H+]⋅([H(n-2)A]2-/[H(n-1)A]-)⋅f2/f1 ................... (2) Kn = [H+]⋅([A]n-/[HA]1-n)⋅fn/fn-1, где K1,K2,...Kn - термодинамические константы диссоциации поликислоты, [H+] - активность ионов водорода, f0,f1,f2,...fn - коэффициенты активности молекулы HnA и ионов H(n_1)A-,H(n-2)A2-,...An-, n - основность кислоты.
Уравнение (2) позволяет определить константы диссоциации поликислот методом потенциометрического титрования при предварительном экспериментальном установлении стандартного потенциала цепи без переноса
Стеклянный электрод /НС1О 4 / AgCl, Ag (I)
в соответствующих растворителях, значение которых E 0 используется для оценки [H + ] в каждой точке титрования поликислоты по уравнению Нернста. Оценка значений коэффициентов активности f i , f 2 , ..., f n проводится по известным методам (например, методом Дэвиса).
Стандартный потенциал цепи (I) определен по методике В.В.Александрова с сотр. [3], а также методом параболического приближения функции
E’ = (Е + 2 6 lgm a ) при m a = 0. (3)
Математическая обработка результатов эксперимента по компьютерным программам [4.-Приложения II, III] приводит к получению стандартного потенциала Е 0 в среде ДМФ , равного 0.427 B, с коэффициентом корреляции К кор = 0.9300.
При титровании четырехосновной кислоты, например, этилендиаминотетрауксусной кислоты ( ЭДТА ) раствором сильного основания в среде ДМФ , совместно нейтрализуются две карбоксильные группы, затем - остальные две. Проблема определения последовательных термодинамических констант диссоциации электролитов, характеризуемых не только равновесными концентрациями, но и ионной силой и коэффициентами активности всех ионов электролита (а не всего электролита, как зачастую принимается), с точки зрения методологии остается одной из интересных в физической химии растворов сильных и слабых полиэлектролитов.
Новый подход к оценке ионной силы, коэффициентов активности всех равновесных частиц в титруемой системе
При теоретических расчетах и экспериментальных определениях термодинамических констант диссоциации многоосновных кислот ионная сила и коэффициенты активности всех ионов, образующихся при титровании полиэлектролитов, не учитывались. В целях упрощения модельных расчетов, в связи с невозможностью оценки концентрации всех равновесных частиц, в литературе рекомендовалось принимать во внимание изменение концентрации первого иона, образующегося при диссоциации полиэлектролита или при титровании его сильным, однофункциональным электролитом - титрантом [1, 2, 5]. Безусловно, это не может быть оправданно, так как приводит к значительным расхождениям расчетов и эксперимента. Например, коэффициент активности иона водорода при концентрации сильной кислоты С m = 10 моль/1000 г Н 2 О равен 10.54, т.е. расхождение эксперимента от теории столь значительно (более чем в 10 раз), что в уравнение для константы диссоциации хлороводородной кислоты вместо концентрации иона Н + требуется вводить произведение концентрации на коэффициент активности - активность.
В данной работе осуществлен полный учет равновесных концентраций всех частиц в растворе с дальнейшим новым подходом к проблеме ионной силы раствора электролита, заключающимся в графическом представлении логарифмической диаграммы « lg ( активности гидроксония ) - lg С» [6, 7].
Показана возможность и плодотворность такой интерпретации во всем диапазоне изменения концентраций всех равновесных частиц в титруемом растворе при нейтрализации четырехосновной кислоты сильным основанием.
Определение термодинамических констант диссоциации полифункциональных электролитов при совместной нейтрализации сильноосновным титрующим агентом кислотных до сих пор было затруднено вследствие ряда факторов:
-
- невозможности дифференцированной оценки констант в буферных областях в связи с нивелированием «смеси» полиэлектролита;
-
- некорректной оценки ионной силы полиэлектролита, так как при титровании образуется группа трудно рассчитываемых по концентрациям частиц - ионов разной степени зарядности и, как следствие, из-за проблем наиболее приемлемого определения коэффициентов активности всех заряженных частиц, вносящих немалый вклад в расчеты величин термодинамических констант диссоциации.
Применение разработанной нами концепции на основе построенных логарифмических диаграмм дает возможность преодолеть отмеченные трудности .
Для определения термодинамических констант диссоциации многоосновных кислот воспользуемся методами прямой потенциометрии и потенциометрического титрования, как наиболее наглядными и оптимальными среди известных методов. При этом требуется учитывать основные количественные законы теории растворов электролитов : закон электронейтральности, закон сохранения материального баланса, правило стехиометрии и ряд других.
Существующие методы оценок ионной силы при кислотно-основном равновесии, в связи с затруднениями полного и объективного учета концентраций всех образующихся частиц, основаны на использовании концентрации первой ионизированной частицы, например, [ A -] для одноосновной; [ HA - ] для двухосновной; [ H 2 A - ] для трехосновной кислот и т.д. Очевидно, пренебрегать равновесными концентрациями остальных ионов в расчетах констант диссоциации не следует, так как зачастую равновесные концентрации более заряженных частиц в процессе диссоциации значительно превышает концентрации первых ионов. Как уже отмечалось, наиболее приемлемым и плодотворным является представление кривой титрования кислот в логарифмических координатах. Равновесные концентрации неоттит-рованной кислоты [ H 4 A ] и ионов [ Н 3 A -], [ H 2 A 2" ], [ HA 3 ], [ A 4-], образующихся при титровании кислоты
H 4 A , могут быть определены на диаграммах - lg (активность гидроксония) – lgC . Оценка значения коэффициентов активности f 0 , f 1 , f 2 , f 3 , f 4 в уравнении (2) проводится разными методами, например, по закону Дебая [8]:
lg f i = - Az i 2I1/2/( 1 +ВаI1/2), (4)
где A = 1.8246∙106 /(εT)3/2, B = ( 2 e2N A ∙1000)1/2/( εk Б Т)1/2, I – ионная сила раствора, определяемая в данном случае по известному уравнению: I = 0.5([HA-]∙ z 1 2 + [ A2- ]∙ z 2 2), а – расстояние наибольшего сближения ионов.
Обоснование метода Дэвиса при оценке коэффициента активности ионов для определения ионных составляющих в растворах электролитов
При оценке коэффициентов активности наиболее приемлемым является уравнение Дэвиса [9]:
-lgf i = [A∙Z i ∙I0.5/( 1 +I0.5)]- 0.1 ∙I, (5)
так как метод Дебая - уравнение (4) - требует введения а – расстояний наименьшего сближения частиц, которые, строго говоря, являются неопределенными и обычно принимаемыми в пределах 5∙10-8 см . Поскольку в растворе все частицы (даже нейтральные молекулы) находятся в сольватированном состоянии (молекулярные сольваты, сольватированные ионы, ассоциированные ионы и т.д.), то даже простая сумма радиусов сольватированных ионов (в первом приближении) не дает полной информации по расстояниям наименьшего сближения ионов и вряд ли будет постоянной для одних и тех же сольватированных катиона и аниона в разных растворителях и равной 5∙10-8 см [10].
Коэффициент А определяется соотношением
A = ( 2 πN A ∙1000)1/2/( e3/ 2.303 ∙ k Б 3/2) ∙ ( 1 /ε3/2T3/2 ), (6)
где N A – число Авогадро, k Б – постоянная Больцмана, ε – диэлектрическая проницаемость среды, Т – температура по Кельвину. Для ДМФ получена величина А = 1.6363.
Таким образом, уравнение Дэвиса устраняет неопределенность с расстоянием наименьшего сближения частиц и вполне приемлемо для расчетов коэффициентов активности ионов в водных, тем более в неводных, растворах, где в зависимости от природы растворителя одни и те же ионы характеризуются различными радиусами сольватных оболочек. Расчеты термодинамических констант диссоциации электролитов дают более достоверные результаты по сравнению с методом Дебая.
Термодинамические константы четырехосновных кислот в органических растворителях
Концептуальный подход к оценке ионной силы раствора электролита заключался в полном учете концентраций всех равновесных частиц в титруемой системе. Отметим еще раз, что для сложных систем, к которым относится титруемый полиэлектролит, традиционно применялась концентрация только первого диссоциированного иона полиэлектролита, несмотря на присутствие двух- и многозарядных ионов, так как не было предложено реального инструмента, позволяющего количественно оценить продукты диссоциации.
Четырехосновная кислота при нейтрализации функциональных групп с двумя скачками титрования
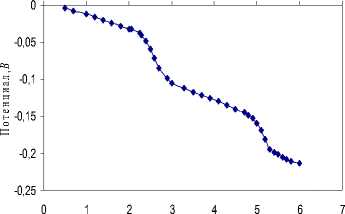
Четырехосновная этилендиаминотетрауксусная ( ЭДТА ) кислота в среде ДМФ титруется с двумя скачками потенциалов, причем первый скачок соответствует совместной нейтрализации первых двух карбоксильных групп, а второй – совместной нейтрализации остальных карбоксильных групп (рис. 1).
В таблице 1 представлены данные потенциометрического титрования 2.3705∙10-3 М раствора ЭДТА 5.04∙10-2 М раствором гуанидина в среде ДМФ, а также -lg a(SH+) , логарифмы равновесных концентраций всех частиц в растворе в процессе титрования, определенных из рисунка 2, их коэффициенты активности, рассчитанные по уравнению (5), рК i исследуемой поликислоты по уравнению (2) для многоосновных кислот.
Объем титранта, мл
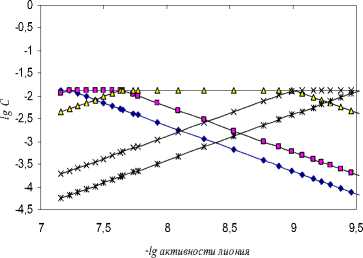
Рис. 2. Логарифмическая диаграмма «lg C – lg активности лиония» ЭДТА в ДМФ ( ◊ - [H 4 A], □ — [H 3 A ], А - [H 2 A2 ], х - [HA3-], * — [A4-])
Рис. 1. Кривая потенциометрического титрования 3.37∙10-3М раствора
ЭДТА 0.0504М раствором гуанидина в ДМФ
Математическая обработка результатов, полученных при потенциометрическом титровании по термодинамическим константам диссоциации ЭДТА в среде ДМФ , приводит к доверительному интервалу, внутри которого с вероятностью 95% находятся показатели констант диссоциации [4.-Прил. IV]:
рК 1 = 7.44 ±0.06; рК 2 = 8.31± 0.18; рК 3 = 11.19 ± 0.19; рК 4 = 12.61 ± 0.26.
Данные для оценки термодинамических констант диссоциации ЭДТА в ДМФ
Таблица 1
|
Vi |
E, В |
-lg a(SH+) |
lg [H 4 A] |
lg[H 3 A-] |
lg[H 2 A2-] |
lg[HA3-] |
lg[A4-] |
|
0.5 |
-0.004 |
7.16 |
-1.8702 |
-1.9309 |
-2.3508 |
-3.7208 |
-4.2508 |
|
0.7 |
-0.008 |
7.23 |
-1.8773 |
-1.8702 |
-2.2830 |
-3.6530 |
-4.1830 |
|
1.0 |
-0.012 |
7.29 |
-1.9451 |
-1.8702 |
-2.2152 |
-3.5852 |
-4.1152 |
|
1.2 |
-0.016 |
7.36 |
-2.0129 |
-1.8702 |
-2.1474 |
-3.5174 |
-4.0474 |
|
1.4 |
-0.020 |
7.43 |
-2.0807 |
-1.8702 |
-2.0796 |
-3.4496 |
-3.9796 |
|
1.6 |
-0.024 |
7.50 |
-2.1485 |
-1.8702 |
-2.0118 |
-3.3818 |
-3.9118 |
|
1.8 |
-0.028 |
7.56 |
-2.2163 |
-1.8702 |
-1.9440 |
-3.3141 |
-3.8441 |
|
2.0 |
-0.032 |
7.63 |
-2.2841 |
-1.8702 |
-1.8763 |
-3.2463 |
-3.7763 |
|
2.2 |
-0.038 |
7.73 |
-2.3857 |
-1.9658 |
-1.8702 |
-3.1446 |
-3.6746 |
|
2.3 |
-0.040 |
7.77 |
-2.4196 |
-1.9997 |
-1.8702 |
-3.1107 |
-3.6407 |
|
2.4 |
-0.049 |
7.92 |
-2.5722 |
-2.1522 |
-1.8702 |
-2.9581 |
-3.4881 |
|
2.5 |
-0.059 |
8.09 |
-2.7417 |
-2.3217 |
-1.8702 |
-2.7886 |
-3.3186 |
|
2.6 |
-0.071 |
8.29 |
-2.9451 |
-2.5251 |
-1.8702 |
-2.5852 |
-3.1152 |
|
2.7 |
-0.085 |
8.53 |
-3.1824 |
-2.7624 |
-1.8702 |
-2.3480 |
-2.8780 |
|
2.9 |
-0.099 |
8.77 |
-3.4196 |
-2.9997 |
-1.8702 |
-2.1107 |
-2.6407 |
|
3.0 |
-0.106 |
8.89 |
-3.5383 |
-3.1183 |
-1.8702 |
-1.9920 |
-2.5220 |
|
3.3 |
-0.112 |
8.99 |
-3.6400 |
-3.2200 |
-1.8702 |
-1.8903 |
-2.4203 |
|
3.5 |
-0.117 |
9.07 |
-3.7247 |
-3.3048 |
-1.9347 |
-1.8702 |
-2.3356 |
|
3.7 |
-0.122 |
9.16 |
-3.8095 |
-3.3895 |
-2.0195 |
-1.8702 |
-2.2508 |
|
3.9 |
-0.126 |
9.23 |
-3.8773 |
-3.4573 |
-2.0873 |
-1.8702 |
-2.1830 |
|
4.1 |
-0.130 |
9.29 |
-3.9451 |
-3.5251 |
-2.1551 |
-1.8702 |
-2.1152 |
|
4.3 |
-0.135 |
9.38 |
-4.0298 |
-3.6098 |
-2.2398 |
-1.8702 |
-2.0305 |
|
4.5 |
-0.140 |
9.46 |
-4.1146 |
-3.6946 |
-2.3246 |
-1.8702 |
-1.9457 |
|
4.7 |
-0.145 |
9.55 |
-4.1993 |
-3.7793 |
-2.4093 |
-1.8793 |
-1.8702 |
Продолжение таблицы 1
|
Vi |
I0.5 |
f 1 |
f 2 |
f 3 |
f 4 |
рК 1 |
рК 2 |
рК 3 |
рК 4 |
|
0.5 |
0.1268 |
0.6568 |
0.4298 |
0.2812 |
0.1840 |
7.40 |
8.13 |
- |
- |
|
0.7 |
0.1367 |
0.6383 |
0.4057 |
0.2579 |
0.1639 |
7.41 |
8.22 |
- |
- |
|
1.0 |
0.1439 |
0.6255 |
0.3893 |
0.2424 |
0.1509 |
7.42 |
8.25 |
- |
- |
|
1.2 |
0.1518 |
0.6117 |
0.3722 |
0.2264 |
0.1378 |
7.43 |
8.28 |
- |
- |
|
1.4 |
0.1607 |
0.5970 |
0.3543 |
0.2103 |
0.1248 |
7.44 |
8.31 |
- |
- |
|
1.6 |
0.1704 |
0.5816 |
0.3360 |
0.1941 |
0.1121 |
7.45 |
8.34 |
- |
- |
|
1.8 |
0.1811 |
0.5653 |
0.3172 |
0.1779 |
0.0998 |
7.46 |
8.38 |
- |
- |
|
2.0 |
0.1929 |
0.5483 |
0.2981 |
0.1621 |
0.0881 |
7.48 |
8.42 |
- |
- |
Продолжение таблицы 1
|
2.2 |
0.1931 |
0.5481 |
0.2978 |
0.1618 |
0.0879 |
7.42 |
8.43 |
- |
- |
|
2.3 |
0.1931 |
0.5481 |
0.2978 |
0.1618 |
0.0879 |
- |
- |
- |
- |
|
2.4 |
0.1950 |
0.5454 |
0.2949 |
0.1594 |
0.0862 |
- |
- |
- |
- |
|
2.5 |
0.2012 |
0.5368 |
0.2855 |
0.1519 |
0.0807 |
- |
- |
- |
- |
|
2.6 |
0.2151 |
0.5187 |
0.2661 |
0.1366 |
0.0701 |
- |
- |
- |
- |
|
2.7 |
0.2421 |
0.4863 |
0.2333 |
0.1119 |
0.0537 |
- |
- |
- |
- |
|
2.9 |
0.2839 |
0.4427 |
0.1924 |
0.0836 |
0.0363 |
- |
- |
- |
- |
|
3.0 |
0.3118 |
0.4176 |
0.1705 |
0.0696 |
0.0284 |
- |
- |
- |
- |
|
3.3 |
0.3399 |
0.3948 |
0.1517 |
0.0583 |
0.0224 |
- |
- |
11.06 |
12.40 |
|
3.5 |
0.3479 |
0.3887 |
0.1469 |
0.0555 |
0.0210 |
- |
- |
11.09 |
12.47 |
|
3.7 |
0.3534 |
0.3847 |
0.1438 |
0.0537 |
0.0201 |
- |
- |
11.12 |
12.50 |
|
3.9 |
0.3601 |
0.3799 |
0.1401 |
0.0516 |
0.0190 |
- |
- |
11.15 |
12.54 |
|
4.1 |
0.3690 |
0.3737 |
0.1353 |
0.0490 |
0.0177 |
- |
- |
11.18 |
12.60 |
|
4.3 |
0.3832 |
0.3641 |
0.1282 |
0.0451 |
0.0158 |
- |
- |
11.24 |
12.68 |
|
4.5 |
0.4011 |
0.3529 |
0.1200 |
0.0408 |
0.0138 |
- |
- |
11.32 |
12.78 |
|
4.7 |
0.4185 |
0.3425 |
0.1127 |
0.0370 |
0.0122 |
- |
- |
11.39 |
12.88 |
Во-первых, доверительный интервал в большинстве обработанных величин более узок, что свидетельствует о достаточно высокой воспроизводимости результатов. Во-вторых, учитываются равновесные концентрации всех частиц в системе в процессе переноса протона. В-третьих, этот метод более нагляден и подтверждается диаграммами. В-четвертых, при решении многих систем уравнений зачастую константы диссоциации оказываются отрицательными.
Таким образом, разработана теоретическая модель определения термодинамических констант диссоциации многоосновных кислот в растворителях и предложена методика экспериментального подтверждения модельных уравнений.
