Изучение генетики болезни фабри в Ленкорань-Астаринском экономическом районе (Азербайджан)
")
Автор: Садыхзаде Н. Н.
Журнал: Бюллетень науки и практики @bulletennauki
Рубрика: Медицинские науки
Статья в выпуске: 9 т.8, 2022 года.
Бесплатный доступ
Целью исследований является изучение генетики болезни Фабри у населения Ленкорань-Астаринского экономического района Азербайджанской Республики. Пациенты были распределены следующим образом: из Астаринского - 16, Масаллинского - 33, Ленкоранского - 17 и Лерикского районов - 10 пациентов. Активность фермента альфа-галактозидаза А и количественное определение глоботриазилсфингозина (Lyso-GB3, Lyso-GL3) определяли методом жидкостной масс-спектроскопии. Ген GLA в образцах ДНК исследовали методом секвенирования нового поколения (NGS). Идентифицирована мутация (NM_000169.2:c.801+3A>G). Согласно «Руководству ACMG*» обнаруженная мутация NM_000169.2:c.801+3A>G по патогенности отнесена к классу 1.
Болезнь фабри, фермент, ген gla, мутация, альфа-галактозидаза, глоботриазилсфингозин (lyso-gb3)
Короткий адрес: https://sciup.org/14125318
IDR: 14125318 | УДК: 616-056.7 | DOI: 10.33619/2414-2948/82/31
Study of fabry disease genetics in Lenkoran-Astara economic region (Azerbaijan)
Goal of our studies is to study genetics of Fabry disease for the population of Lenkoran-Astara Economic Region of Azerbaijan Republic. Patients were from the following areas of the districts: 16 persons from Astara, 33 from Masally, 17 from Lenkoran and 10 from Lerik. Alpha-galactosidase A enzyme activity and globotriasylsfingosine (lyso-Gb3, Lyso-GL3) quantitative identification were done with liquid mass-spectroscopy method. GLA gene in DNA samples was studied with new generation sequencing (NGS). Identified mutation is (NM_000169.2:c.801+3A>G). According to the Guidelines of ACMG* mutation identified NM_000169.2:c.801+3A>G is classified as pathogenic class 1.
Текст научной статьи Изучение генетики болезни фабри в Ленкорань-Астаринском экономическом районе (Азербайджан)
Бюллетень науки и практики / Bulletin of Science and Practice
УДК 616-056.7
Впервые в Ленкорань-Астаринском регионе Азербайджанской Республики проведены популяционно-генетические исследования по выявлению и изучению генетики болезни Фабри. Для скринига пациентов с болезнью Фабри использовали метод определения активности фермента альфа-галактозидаза А и количественного определения глоботриазилсфингозина методом жидкостной масс-спектроскопии. У больных по клиническим показаниям был произведен забор крови на DBS карты (Dry Blood Sample). Обследованные пациенты были распределены следующим образом: из Астаринского района -16, Масаллинского - 33, Ленкоранского - 17 и Лерикского - 10 пациентов. Уровень дефицита фермента альфа-галактозидазы А у гемизиготных мужчин варьировал в пределах 0,0
мкмол/л/ч-2,0 мкмол/л/ч, в среднем 1,0 мкмол/л/ч (Н ≥ 15,3 мкмoл/л/ч). Количество Lyso-Gb3 было характерно повышено для болезни Фабри и варьировало в пределах 106,0нг/мл-218,0 нг/мл, при среднем значении 112,2 нг/мл (Н≤ 1,8). Что касается гетерозиготных женшин, то активность фермента альфа-галактозидазы А также была снижена и варьировала в вределах 2,0 мкмол/л/ч-4,3 мкмол/л/ч, в среднем 3,67 мкмол/л/ч (Н≥ 15,3 мкмoл/л/ч). Следовательно, наблюдали повышенные значения Lyso-Gb3 варьирующие в пределах 8,3 нг/мл-20,0 нг/мл, при среднем значении 15,0 нг/мл (Н≤ 1,8). Клинико-генеологический анализ, проведенный у членов семьи пробанда Т.И. из Масаллинского района, дополнительно установил семь женщин и троих мужчин с подозрением на болезнь Фабри. В большой семье из 28 человек выявлено 12 больных с диагнозом болезнь Фабри, внутрисемейная частота которой составила 42,86%. Идентификацию мутации гена GLA проводили методом секвенирования нового поколения (NGS). Идентифицирована мутация интрона 5 (NM_000169.2: c.801+3A>G). Согласно «Руководству ACMG*» идентифицированная нами мутация по патогенности относится к 1 классу, в соответствие в Рекомендациями Centogene.
Болезнь Фабри (БФ), ангиокератома (Corporis diffusum), болезнь Андерсона-Фабри редкое генетически детерминированное заболевание с Х-сцепленным типом наследования, из группы лизосомных болезней накопления [1, 2].
В 1898 году Фабри описал 13-летнего мальчика с нодулярной пурпурой, у которого, в последствии, развилась альбуминурия. Данный клинический случай был классифицирован автором как один из вариантов диффузной ангиокератомы. В том же году Андерсон описал 39-летнего мужчину с ангиокератомой, протеинурией, деформациями пальцев рук, варикозным расширением вен и лимфатическим отеком [3-6].
Причиной возникновения БФ являются мутации гена GLA, кодирующего фермент альфа-галактозидазу А (GLA; EC 3.2.1.22). Ген GLA картирован на длинном плече хромосомы Хq22.1, имеет размер около 12 тысяч пар нуклеотидов и состоит из 7 экзонов. К настоящему времени идентифицировано более 600 вариантов в гене GLA, в том числе около 500 патогенных мутаций, изменяющих свойства и стабильность альфа -галактозидазы А [7, 8].
Большинство мутаций являются уникальными для каждой семьи. БФ. Первичным биохимическим дефектом при БФ является недостаточность фермента альфа-галактозидаза А, который отщепляет терминальный остаток α-галактозы олигосахаридной цепи нейтральных гликосфинголипидов. Недостаточность фермента приводит к накоплению в лизосомах разных клеток (эндотелиальные и гладкомышечные клетки сосудов, эпителиальные клетки большинства органов, центральной нервной системы, сердца) гликосфинголипидов [9-11].
При болезни Фабри наблюдается нарушение потоотделения, быстрая утомляемость и непереносимость физических нагрузок, ангиокератомы, воронковидная кератопатия, нарушения функции сердца, почек, мозга и нервной системы, проблемы в психоэмоциональной сфере. На современном этапе накоплено достаточно данных, чтобы считать тип наследования болезни Фабри X-сцепленным доминантным с неполной пенетрантностью у женщин [12, 13].
Распространенность наследственной болезни Фабри составляет от 1 на 40 000 до 1 на 120 000 живых новорожденных, и является одной из наиболее распространенных лизосомных болезней накопления после болезни Гоше. Встречается во всех расовых группах и возникает с частотой 1:117000 в Австралии, 1:476000 в Нидерландах, 1:40000-60000 мужчин в США [14-16].
Следует с сожалением отметить, что до недавнего времени болезнь Фабри, из-за сложности диагностики, недостаточно изучена среди больных у населения Республики.
Следовательно, сегодня из-за доступности молекулярно-генетических методов диагностики, целью наших иследований является изучение генетики болезни Фабри у населения Ленкорань-Астаринского региона Азербайджанской Республики.
Материал и методы
Экспериментальный материал собран во время экспедиционных работ в Центральных районных больницах Астаринского, Ленкоранского, Масаллинского и Лерикского районов, расположенных в юго-восточной части Азербайджанской Республики. У 76 больных по клиническим показаниям был произведен забор крови на DBS карты (Dry Blood Sample) и отправлены в лабораторию для дальнейших генетических исследований.
Пациенты были распределены следуюшим образом: 16 человек из Астаринского района, 33 из Масаллинского, 17 из Ленкоранского и 10 пациентов из Лерикского районов. Высушенные пятна крови очень удобны для скрининговых исследований в экспедиционных условиях, так как сухие пятна крови легко получить, хранить и транспортировать в лабораторию. В дальнейших исследованиях использовали также жидкую кровь (в пробирке с гепарином или ЭДТА) .
Активность фермента альфа-галактозидазу А определяли методом жидкостной масс-спектроскопии. Для уточнения диагноза нами был использован новый тест лля количественного определения глоботриазилсфингозина (Lyso-GB3, Lyso-GL3) более наглядно, нежели глоботриазилцерамида (GB3, GL3) [17].
Ген GLA в образце ДНК, полученном из взятой периферической крови пациента, исследовали методом секвенирования нового поколения. «Более 99% кодирующих областей этих генов были изучены с глубиной чтения не менее 50X. Средняя глубина чтения составляет 1559 показаний. В анализ были включены соединения экзон-интрон (±10 п.н.). Классификацию патогенности полученных данных проводили согласно «Руководству ACMG*».
Результаты и их обсуждение
Генетический скрининг активности фермента альфа-галактозидазы 76 больных с диагнозом кардиомиопатия выявил шесть пациентов с дефицитом данного фермента, из которых 4 мужчины и 2 женщины. Уровень активности альфа-галактозидазы у мужчин был значительно снижен и варьировал в пределах 0,0 мкмол/л/ч-0,8 мкмол/л/ч при норме ≥ 15,3 мкмол/л/ч. Для подтверждения диагноза использован тест Lyso-Gb3, что подтвердил ранее поставленный диагноз ферментным анализом. Уровни Lyso-Gb3 у мужчин варьировали в предах 106,0 нг/мл-117,0 нг/мл при норме ≤ 1,8 нг/мл. При болезни Фабри активность альфа-галактозидазы в крови у мужчин всегда снижена.
У женщин уровень активности фермента альфа-галактозидазы был занижен и составлял 1,2 мкмол/л/ч и 1,9 мкмол/л/ч, соответственно. Также наблюдали повышение количества Lyso-Gb3 8,3-15,6 нг/мл при норме ≤ 1,8 нг/мл, что подтверждает поставленный диагноз.
Использование теста Lyso-Gb3 позволяет не только разъяснить трудные диагностические случаи болезни, но и определить классическую или неклассическую форму заболевания, а также для мониторинга состояния пациента [17-18].
Трое пациентов — двое мужчин и одна женщина были выявлены в Масаллинском районе. Все трое больных были представителями одной большой семьи. Двое пациентов (мужчина и женщина) из Астаринского и один мужчина из Ленкоранского районов. Клинико- генеалогический анализ членов семей больных пациентов позволил нам дополнительно установить семь женщин и троих мужчин с подозрением на болезнь Фабри. Для всех членов семей с подозрением на болезнь Фабри была взята кровь и проведен анализ на активность фермента альфа-галактозидазы и количественный анализ на Lyso-Gb3.
Результаты ферментного и Lyso-Gb3 анализов семьи Т.И. из Масаллинского района представлены в Таблице.
Таблица
РЕЗУЛЬТАТЫ ФЕРМЕНТНОГО И LYSO-GB3. АНАЛИЗОВ У ЧЛЕНОВ СЕМЬИ ПРОБАНДА Т.И.
|
Пациенты |
Дата рождения |
Пол |
Альфа-галактозидаза Н ≥15.3 мкмол/л/ч |
lyso-Gb3 Н≤ 1,8. нг/мл |
Мутация гена GLA |
Генотип |
|
T.И. |
03.06.87 |
муж |
0.8 мкмол/л/ч |
106.0 нг/мл |
801+3A>G |
Гемизигота |
|
T.A. |
21.06.86 |
муж |
0.8 мкмол/л/ч |
117.0 нг/мл |
801+3A>G |
Гемизигота |
|
T.Г |
19.08.62 |
жен |
3.2 мкмол/л/ч |
15.6 нг/мл |
801+3A>G |
Гетерозигота |
|
A.Г. |
06.09.56 |
жен |
2.0 мкмол/л/ч |
8.3 нг/мл |
801+3A>G |
Гетерозигота |
|
M.А. |
20.07.12 |
муж |
2.0 мкмол/л/ч |
109.0 нг/мл |
801+3A>G |
Гемизигота |
|
K.Э. |
16.08.75 |
жен |
3.0 мкмол/л/ч |
13.0 нг/мл |
801+3A>G |
Гетерозигота |
|
K.A. |
28.07.07 |
жен |
4.0 мкмол/л/ч |
11.0 нг/мл |
801+3A>G |
Гетерозигота |
|
Л.И. |
31.08.51 |
жен |
4,1 мкмол/л/ч |
19.0 нг/мл |
801+3A>G |
Гетерозигота |
|
K.С. |
12.06.78 |
муж |
0.0 мкмол/л/ч |
218.0 нг/мл |
801+3A>G |
Гемизигота |
|
K.Л. |
20.07.53 |
муж |
1.4 мкмол/л/ч |
211.0 нг/мл |
801+3A>G |
Гетерозигота |
|
A.A. |
29.05.17 |
жен |
4.3 мкмол/л/ч |
20.0 нг/мл |
801+3A>G |
Гетерозигота |
|
K.Ф. |
15.05.03 |
жен |
4.1 мкмол/л/ч |
18.0 нг/мл |
801+3A>G |
Гетерозигота |
Уровень дефицита фермента альфа-галактозидазы у гемизиготных мужчин варьировал в пределах 0,0 мкмол/л/ч-2,0 мкмол/л/ч и в среднем составил 1,0 мкмол/л/ч (Н ≥ 15,3 мкмол/л/ч). Количество Lyso-Gb3 было характерно для болезни Фабри значительно повышено и варьировало в пределах 106,0нг/мл-218,0 нг/мл, пр и среднем значении 112,2 нг/мл (Н≤1,8).
Что касается гетерозиготных женщин, активность фермента альфа-галактозидазы также была снижена и варьировала в пределах 2,0 мкмол/л/ч-4,3 мкмол/л/ч, в средней 3,67 мкмол/л/ч (Н ≥ 15,3 мкмол/л/ч). Следовательно, наблюдали повышенные значения Lyso-Gb3, варьирующие в пределах 8,3 нг/мл-20,0 нг/мл, при среднем значении 15,0 нг/мл
При болезни Фабри активность альфа-галактозидазы в крови у мужчин всегда снижена, а (Н≤1,8).у женщин активность фермента может быть около нижней границы нормы, чуть ниже ее или нормальной, из-за чего ферментный анализ для женщин не показателен в отношении наличия/отсутствия болезни Фабри. Следует отметить, что в наших исследованиях у всех женщин-гетерозигот уровень активности фермента был значительно снижен.
Следуюшим этапом наших исследований была молекулярная диагностика гена GLA у всех членов семьи с биохимически установленным диагнозом болезни Фабри.
Образцы ДНК, полученные из высушенных пятень крови (DBS карт), а также из периферической крови больных, исследовали методом секвенирования нового поколения (NGS). «Более 99% кодирующих областей этих генов были изучены с глубиной чтения не менее 50X. Средняя глубина чтения составляло 1559 показаний. В анализ были включены соединения экзон-интрон (±10 п.н.). Классификацию патогенности полученных данных проводили согласно «Руководству ACMG*». Для всех испытуемых идентфицирована мутация GLA гена. Замена трех нуклеотидов аденини на нуклеотид гуанин в 801 позиции интрона 5 (NM_000169.2:c.801+3A>G). Согласно «Руководству ACMG*», идентифицированная нами мутация NM_000169.2:c.801+3A>G по патогенности относится к классу 1 [19].
Мужчины имели гемизиготный генотип, все женщины являлись гетерозиготными носителями данной мутации. Результаты молекулярно-генетических анализов представлены в таблице №1.
Вариант GLA c.801+3A>G предварительно заявлен как разрушающий высоко законсервированный донорский сплайс сайт интрона 5. В соответствие с HGMD Professional 2019.1, этот вариант предварительно описан авторами Shabbeer et al., 2006 (PMID: 16595074) как вызывающий заболевание Фабри. В списке ClinVar он есть и обозначен как патогенный (в клиническом тестировании значится как Variation ID: 197639). Он классифицируется как патогенный (класс 1) в соответствие в Рекомендациями Centogene и ACMG.
Составлена большая родословная для семьи Т.И., объединяющая 28 членов семьи в четырех поколениях, в которой 12 (5 мужчин и 7 женщин) имели болезнь Фабри (Рисунок). Болезнь Фабри, внутрисемейная частота которой составила 42,86%.
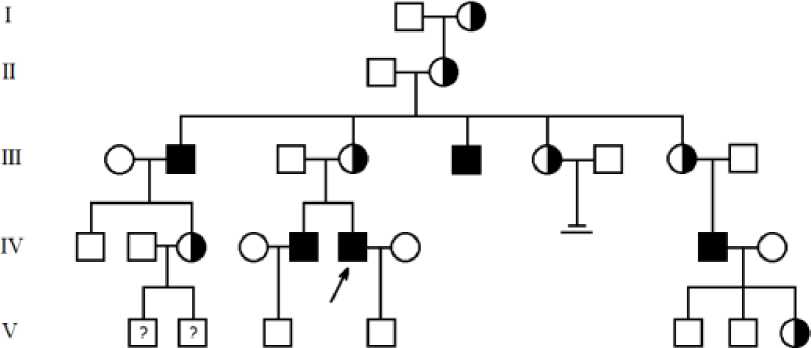
Рисунок. Родословная семьи Т.И.
Таким образом, для изучения генетики болезни Фабри у населения Ленкорань-Астаринского региона Азербайджанской Республики нами собран экспериментальный материал во время экспедиционных работ в Центральных районных больницах Астаринского, Ленкоранского, Масаллинского и Лерикского районов. У 76 больных по клиническим показаниям и с диагнозом кардиомиопатия был произведен забор крови на DBS карты. На основании активности фермента альфа-галактозидазы и количества Lyso-Gb3 в Масаллинского районе у 12-ти пациентов выявлена болезнь Фабри. Выявлено по двое больных из Ленкоранского и Астаринского районов. Для больных из Масаллинкого района идентфицирована мутация GLA гена. Замена трех нуклеотидов аденини на нуклеотид гуанин в 801 позиции интрона 5 (NM_000169.2:c.801+3A>G). Согласно «Руководству ACMG*» идентифицированная нами мутация NM_000169.2:c.801+3A>G по патогенности относится к классу 1. Обсуждаются пути профилактики болезни Фабри для семей репродуктивного возраста.
Выводы
-
1. Впервые в Азербайджанской Республике в большой семье из 28 человек выявлено 12 больных с диагнозом Болезнь Фабри и изучена генетика гена GLA.
-
2. Диагностику Болезни Фабри следует проводить с определения активности фермента альфа-галактозидаза и количественного определения глоботриазилсфингозина. При этом активность фермента альфа-галактозидаза бывает ниже, а количество глоботриазилсфингозина выше нормы.
-
3. Идентфицирована замена трех нуклеотидов аденин на нуклеотид гуанин в 801 позиции интрона 5 (NM_000169.2:c.801+3A>G). Согласно «Руководству ACMG*»,
-
4. Обсуждаются пути профилактики болезни Фабри в виде пренатальной диагностики для семей с генетическим риском.
идентифицированная нами мутация по патогенности относится к классу 1, в соответствие в Рекомендациями Centogene и ACMG.
Список литературы Изучение генетики болезни фабри в Ленкорань-Астаринском экономическом районе (Азербайджан)
- Волгина С. Я. Болезнь Фабри // Практическая медицина. 2012. №7 (62). С. 75-79.
- Волгина С. Я., Асанов А. Ю., Соколов А. А. Диагностика и лечение болезни Фабри у детей раннего возраста // Российский педиатрический журнал. 2015. Т. 18. №6. С. 41-45.
- First CE-IVD assay for lyso-GL-3. https://goo.su/aYhslU
- Sigmundsdottir L. et al. Cognitive and psychological functioning in Fabry disease // Archives of clinical neuropsychology. 2014. V. 29. №7. P. 642-650. https://doi.org/10.1093/arclin/acu047
- Moore D. F. et al. Increased signal intensity in the pulvinar on T1-weighted images: a pathognomonic MR imaging sign of Fabry disease // American Journal of Neuroradiology. 2003. V. 24. №6. P. 1096-1101.
- Smid B. E. et al. Plasma globotriaosylsphingosine in relation to phenotypes of Fabry disease // Journal of medical genetics. 2015. V. 52. №4. P. 262-268. http://dx.doi.org/10.1136/jmedgenet-2014-102872
- Kobayashi M. et al. Frequency of de novo mutations in Japanese patients with Fabry disease // Molecular Genetics and Metabolism Reports. 2014. V. 1. P. 283-287. https://doi.org/10.1016/j.ymgmr.2014.07.001
- Kurschat C. E. Fabry disease—what cardiologists can learn from the nephrologist: a narrative review // Cardiovascular Diagnosis and Therapy. 2021. V. 11. №2. P. 672. https://doi.org/10.21037%2Fcdt-20-981
- Odom R. B. et al. Andrews' diseases of the skin: clinical dermatology. Philadelphia: WB Saunders Company, 2000. V. 1135.
- Pisani A. et al. The kidney in Fabry's disease // Clinical genetics. 2014. V. 86. №4. P. 301309. https://doi.org/10.1111/cge.12386
- Barac I. S. Fabry disease // Romanian Journal of Neurology. 2018. V. 17. №4. P. 200. https://doi.org/10.37897/RJN.2018A5
- Sigmundsdottir L. et al. Cognitive and psychological functioning in Fabry disease // Archives of clinical neuropsychology. 2014. V. 29. №7. P. 642-650. https://doi.org/10.1093/arclin/acu047
- Young-Gqamana B. et al. Migalastat HCl reduces globotriaosylsphingosine (lyso-Gb3) in Fabry transgenic mice and in the plasma of Fabry patients // PLoS One. 2013. V. 8. №3. P. e57631. https://doi.org/10.1371/journal.pone.0057631
- van Breemen M. J. et al. Reduction of elevated plasma globotriaosylsphingosine in patients with classic Fabry disease following enzyme replacement therapy // Biochimica et Biophysica Acta (BBA)-Molecular Basis of Disease. 2011. V. 1812. №1. P. 70-76. https://doi.org/10.1016/j.bbadis.2010.09.007
- Yousef Z. et al. Left ventricular hypertrophy in Fabry disease: a practical approach to diagnosis // European Heart Journal. 2013. V. 34. №11. P. 802-808. https://doi .org/10.1093/eurheartj/ehs166
- Arning K. et al. FabryScan: a screening tool for early detection of Fabry disease // Journal of neurology. 2012. V. 259. №11. P. 2393-2400. https://doi.org/10.1007/s00415-012-6619-y
- Burlina A. P. et al. The pulvinar sign: frequency and clinical correlations in Fabry disease // Journal of neurology. 2008. V. 255. №5. P. 738-744. https://doi.org/10.1007/s00415-008-0786-x
- Ronald B. J. et al. Fabry Disease:: Renal Sonographic and Magnetic Resonance Imaging Findings in Affected Males and Carrier Females With the Classic and Cardiac Variant Phenotypes // Journal of computer assisted tomography. 2004. V. 28. №2. P. 158-168.
- Shabbeer J. et al. Fabry disease: Identification of 50 novel a-galactosidase A mutations causing the classic phenotype and three-dimensional structural analysis of 29 missense mutations // Human genomics. 2006. V. 2. №5. P. 1-13. https://doi.org/10.1186/1479-7364-2-5-297
