Изучение геометрических, термодинамических свойств и гибкости углеводородных молекул методом Монте-Карло

Автор: Журкина Дмитрий Викторович, Рабинович Лександр Львович
Журнал: Ученые записки Петрозаводского государственного университета @uchzap-petrsu
Рубрика: Физико-математические науки
Статья в выпуске: 8 (145) т.1, 2014 года.
Бесплатный доступ
Одна из главных задач физики конденсированного состояния - изучение взаимосвязей между химическим строением и физическими свойствами разных молекул. Углеводородные цепные молекулы играют важную роль в природных системах, широко используются в областях технологии. В настоящей работе методом Монте-Карло проведено моделирование 65 цепных углеводородных молекул вида CH-(CH 2) a-(CH=CH-CH 2) d-(CH 2) b-CH 3 (где a, b, d - целые). Изучены варианты N = 16, 18, 20, 22 (где N = a + b + 3d + 2 - количество атомов углерода), d = 0,1,..., 6 - количество двойных связей (конфигурация cis-); температура T = 293, 303 и 313 K. Все исследованные молекулы рассматривали в невозмущенном состоянии, генерирование значений торсионных углов осуществляли методом существенной выборки в диапазоне 0-360° и учетом взаимозависимости каждых трех из них вдоль по цепи. В итоге моделирования для каждой молекулы вычислены равновесная гибкость, конформационная теплоемкость, относительные флуктуации квадрата радиуса инерции и квадрата расстояния между концевыми атомами углерода. Проанализированы зависимости этих свойств от параметров строения молекул. Обнаружен ряд закономерностей, в том числе корреляция между величиной гибкости и относительными флуктуациями геометрических размеров молекул. Предложена интерпретация полученных зависимостей на основе данных эксперимента о характеристиках внутреннего вращения в цепях данного вида. Полученные данные способствуют углублению общего понимания взаимосвязей между структурой и свойствами рассмотренных молекул.
Ненасыщенные углеводороды, метод монте-карло, равновесная гибкость, флуктуации, конформационная теплоемкость
Короткий адрес: https://sciup.org/14750795
IDR: 14750795 | УДК: 541.64:539.199
Monte Carlo study of geometric, thermodynamic properties and flexibility of hydrocarbon chain molecules
One of the main problems of condensed matter physics is investigation of interconnections between chemical structures and physical properties of various molecules. The hydrocarbon chain molecules play an important role in natural systems. They are widely used in different fields of technology. In this paper, Monte Carlo simulation of 65 hydrocarbon chain molecules CH3-(CH2) a-(CH=CH-CH2)d-(CH2)b-CH3 (where a, b, d are integer) was carried out. Sets of N = 16, 18, 20, 22 (where N = a + b + 3d + 2 is carbons number), d = 0, 1,..., 6 (where d is cis double bonds number) are studied; temperature T = 293, 303 and 313 K. All studied molecules were treated in unperturbed state, torsion angles were generated using importance sampling technique in 0 - 360 deg range and three-wise interdependence of torsions along the chain. As a result, the equilibrium flexibility, conformational heat capacity, relative fluctuations of square radius of gyration, and square end-to-end distance of each molecule were calculated in the simulations. Dependencies of the properties on the molecule structure parameters were analyzed. The set of regularities, including correlations between flexibility and relative fluctuations of geometric dimensions, were found. An interpretation of obtained dependencies based on experimental data of characteristics of internal rotation in the chains, was proposed. The obtained data provide further insight into interconnections between the structure and properties of considered molecules.
Текст научной статьи Изучение геометрических, термодинамических свойств и гибкости углеводородных молекул методом Монте-Карло
Одной из фундаментальных проблем физики конденсированного состояния является установление взаимосвязей между химическим строением и физическими свойствами разных молекул и молекулярных систем в различных условиях. Компонентами таких систем часто выступают молекулы цепного строения [6]. Типичный пример – углеводородные цепные молекулы. Информация об их физических и химических свойствах важна не только с научной точки зрения, но и для развития разных технологических отраслей, причем обе задачи тесно связаны между собой. А именно, обсуждаемые данные нужны для углубления знаний о структуре и функциях биологических систем, так как углеводородные цепи входят в состав молекул фосфолипидов мембран [1], [16]. С другой стороны, существование последних основано на эффектах самоорганизации, а принцип самоорганизации является одним из базовых принципов современных нанотехнологий. Несмотря на острую необходимость в данных по свойствам многих систем, они подчас скудны или
отсутствуют в литературе. В настоящей работе методом Монте-Карло (МК), алгоритм которого был разработан ранее [5], при температурах T = 293, 303, 313 K проведено моделирование 65 углеводородных цепных молекул с двойными связями cis , вида CH3 – (CH2) a – (CH=CH – CH2)d – – (CH2) b – CH3 (где a , b , d – целые), в невозмущенном состоянии (в Θ-условиях [6]), с количеством N атомов углерода (где N = a + b + 3d + 2), равным 16, 18, 20, 22, и количеством двойных связей d = 0, 1, 2, ..., 6. В работе [3] при моделировании перечисленных выше молекул этим методом были исследованы характеристики их формы, а в настоящей работе изучены равновесная гибкость, конформационная теплоемкость, относительные флуктуации квадрата радиуса инерции и квадрата расстояния между концевыми атомами углерода молекул.
Модель цепной молекулы и расчет средних характеристик
«Θ-условия», при которых вычисляли свойства всех молекул, примерно отвечают условиям в жидком или аморфном состояниях вещества [6], [10], [22]. Основные этапы расчета состояли в следующем (математические основы модели и алгоритма МК описаны в [3], [5]). Моделировали конформационное поведение одиночных цепей, среднее значение
где R c – радиус-вектор центра масс рассматриваемой цепи:
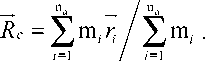
Здесь m
.
(i
=1, 2, .„, n
a
) - массы атомов;
^
(где
i
= 1, 2, …, n
a
) – радиус-векторы атомов в данной конформации; n
a
– общее количество атомов. Вычислены средние квадраты расстояний
^ 2 = « S 4) Ч S 21 )/( S 2)2’
е =(( 4ЧИ)/Ч’>.
Изучена равновесная гибкость молекул; в качестве меры гибкости использовано отношение
Описание строения молекул
Полное описание строения любой из рассматриваемых здесь углеводородных молекул требует указания общего количества атомов углерода (N) в ней, количества двойных связей (d), а также их положения в цепи. Последнее в общем случае требует указания еще d чисел. Однако перечислять положения каждой двойной связи не обязательно, поскольку химическое строение этих молекул специфично : между каждой парой двойных связей расположена только одна группа CH2, и положение всех двойных связей легко вычисляется, если известно, например, местоположение только первой двойной связи. Для этого можно задать параметр Δ [17] – номер атома углерода, ближайшего к заданному концу цепи и участвующего в образовании первой (от данного конца цепи) двойной связи. Атомы углерода при этом имеют номера от 1 до N подряд вдоль по цепи, начиная от одного из концевых. Положение всех двойных связей с помощью одного параметра можно описать и другим способом: указать местоположение X их «центра» [17], которое равно среднему арифметическому номеров атомов углерода, участвующих в образовании всех двойных связей. Например, если молекула содержит одну двойную связь, расположенную между 9-м и 10-м атомами углерода, то Δ = 9, а X = (9 + 10)/2 = 9,5; если молекула содержит 2 двойные связи, расположенные между 6-м и 7-м, 9-м и 10-м атомами углерода, то Δ = 6, а X = (6 + 7 + 9 + 10)/4 = 8, и т. д. В зависимости от обстоятельств в настоящей работе использован 1-й или 2-й вариант идентификации молекул (N, d, Δ или N, d, X). Параметры N, d, Δ в обозначениях молекул указаны сокращенной формулой N:d w ^cis.
РЕЗУЛЬТАТЫ И ОБСУЖДЕНИЕ
Численные значения ряда полученных в настоящей работе характеристик (<
S
>, <
S
2>,
-
2>) для некоторых молекул и соответствующие данные литературы [8], [9], [10], [12], [13], [15], [17], [18], [19], [20] представлены в таблице; в целом они согласуются между собой. Важно отметить, что в большинстве работ приведены данные лишь для отдельных молекул или неболь- - ших групп, а в настоящей работе в идентичных условиях исследована большая их совокупность (65 молекул). Это позволяет выявить взаимосвязь между химическим строением молекул и свойствами веществ. При этом следует учесть, что на результаты могут оказывать влияние избранная
Характеристики* размеров углеводородных цепных молекул, полученные в эксперименте или при компьютерном моделировании (в расплаве или Θ-условиях). Параметры N, d, Δ в обозначениях молекул указаны сокращенной формулой N:dω Δcis
|
Молекула |
T , K |
< S >, Å |
(< S 2>)1/2, Å |
< S 2>, Å2 |
|
( |
|
Метод** |
Ссылка |
|
16:0 |
298 |
22±2 |
НР |
[10] |
|||||
|
298 |
4.9±0.2 |
МК |
[12]*** |
||||||
|
298 |
26.02±0.03 |
226.5±0.4 |
МД-а |
[15] |
|||||
|
298 |
23.10±0.04 |
189.3±0.6 |
МД-б |
[15] |
|||||
|
303 |
5.04±0.01 |
14.67±0.01 |
МК |
[8] |
|||||
|
303 |
4.909±0.001 |
24.26±0.01 |
13.95±0.01 |
199.9±0.1 |
МК |
Наст. раб. |
|||
|
313 |
4.903±0.001 |
24.20±0.01 |
13.92±0.01 |
199.2±0.1 |
МК |
Наст. раб. |
|||
|
318 |
4.8±0.2 |
НР |
[13] |
||||||
|
323 |
25.34±0.02 |
217.5±0.2 |
МД-а |
[15] |
|||||
|
323 |
22.66±0.02 |
183.7±0.3 |
МД-б |
[15] |
|||||
|
373 |
24.49±0.02 |
206.8±0.3 |
МД-а |
[15] |
|||||
|
373 |
21.94±0.02 |
175.1±0.3 |
МД-б |
[15] |
|||||
|
18:0 |
278 |
5.10±0.01 |
26.54±0.02 |
14.16±0.01 |
214.6±0.2 |
МК |
[17], [18]***** |
||
|
298 |
5.3±0.1 |
МК |
[12]*** |
||||||
|
303 |
5.05±0.01 |
26.06±0.01 |
13.95±0.01 |
208.8±0.2 |
МК |
[17],[18]***** |
|||
|
303 |
5.391±0.001 |
29.30±0.01 |
15.28±0.01 |
241.3±0.1 |
МК |
Нас т. раб. |
|||
|
313 |
5.384±0.001 |
5.406±0.001 |
29.22±0.01 |
15.25±0.01 |
15.51±0.01 |
240.5±0.1 |
МК |
Наст. раб. |
|
|
323 |
15.84±0.07 |
МК |
[20]**** |
||||||
|
497.5 |
4.92±0.01 |
13.52±0.07 |
БД |
[19] |
|||||
|
18:1 to 9cis |
313 |
5.010±0.001 |
14.09±0.01 |
МК |
Наст. раб. |
||||
|
497.5 |
4.80±0.01 |
13.18±0.04 |
БД |
[19] |
|||||
|
18:2 to 6cis |
278 |
4.52±0.01 |
20.91±0.1 |
11.99±0.04 |
157.9±0.8 |
МК |
[17], [18]***** |
||
|
313 |
4.745±0.001 |
4.769±0.001 |
22.74±0.01 |
12.92±0.01 |
13.31±0.01 |
177.1±0.1 |
МК |
Наст. раб. |
|
|
497.5 |
4.47±0.01 |
11.66±0.05 |
БД |
[19] |
|||||
|
18:3 to 3cis |
278 |
4.39±0.04 |
19.73±0.3 |
11.34±0.2 |
141.2±3.0 |
МК |
[17], [18]***** |
||
|
313 |
4.655±0.001 |
4.678±0.001 |
21.88±0.01 |
12.59±0.01 |
12.94±0.01 |
167.4±0.2 |
МК |
Наст. раб. |
|
|
497.5 |
4.32±0.01 |
10.60±0.05 |
БД |
[19] |
|||||
|
18:4 to 3cis |
278 |
4.10±0.02 |
17.20±0.2 |
10.16±0.1 |
116.0±2.0 |
МК |
[17], [18]***** |
||
|
313 |
4.446±0.001 |
19.94±0.01 |
11.91±0.01 |
150.3±0.2 |
МК |
Наст. раб. |
|||
|
18:5 to 3cis |
278 |
3.96±0.01 |
16.06±0.01 |
9.46±0.01 |
101.4±0.2 |
МК |
[17], [18]***** |
||
|
303 |
3.95±0.01 |
16.05±0.01 |
9.47±0.01 |
101.5±0.2 |
МК |
[17], [18]***** |
|||
|
313 |
4.369±0.002 |
19.26±0.02 |
11.60±0.02 |
141.9±0.4 |
МК |
Наст. раб. |
|||
|
20:0 |
298 |
5.7±0.1 |
МК |
[12]*** |
|||||
|
303 |
5.856±0.001 |
34.61±0.01 |
284.1±0.1 |
МК |
Наст. раб. |
||||
|
313 |
5.848±0.001 |
34.52±0.01 |
283.0±0.1 |
МК |
Наст. раб. |
||||
|
318 |
5.1±0.3 |
НР |
[13] |
||||||
|
500 |
36.17±0.01 |
312.7±0.1 |
МК |
[9] |
|||||
|
500 |
36.18±0.01 |
313.6±0.1 |
МК |
[9] |
|||||
|
500 |
36±1 |
311±15 |
МД |
[9] |
0240240240246 d
Рис. 2. Зависимости отношений
8 ( ), 9 ( ), 11 ( ) по группам молекул с одина ковым количеством N атомов углерода. Параметр Δ – номер атома углерода, участвующего в образовании первой двойной связи от конца цепи. Расчет методом МК при температуре T = 293 K. Доверительные интервалы, отвечающие 95%-ной надежности согласно t-распределению Стьюдента, меньше размеров символов на графиках d = Const и N = Const, тем, как правило, больше ее гибкость (см. рис. 1). Этот эффект объясняется разной степенью влияния наиболее низкоэнергетических (свернутых) конформаций фрагментов, содержащих двойные связи. Если фрагмент с двойными связями расположен у конца цепи, свернутые конформации оказываются в известном смысле «локальными» по сравнению с вытянутыми низкоэнергетическими конформациями оставшегося (насыщенного) участка цепи. Нали- чие такого фрагмента вблизи середины цепи разделяет насыщенную часть молекулы на 2 участка, низкоэнергетические конформации которых в общем случае вытянуты в разных направлениях, что уменьшает расстояние
Конкурентное влияние на величину
Наконец, при d = Const и X = Const гибкость молекулы растет (
Обсуждаемые для
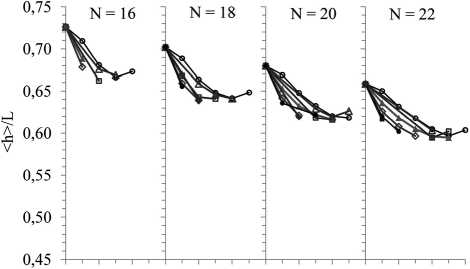
Относительные флуктуации геометрических размеров
. Наблюдается рост флуктуаций
E
h
2
(рис. 3) для всех моноеновых и диеновых цепей (d = 1, 2), а также достаточно длинных (N = 18, 20, 22) триеновых и тетраеновых цепей (d = 3, 4) при смещении X от концов к середине цепи. Если количество двойных связей в цепи еще больше (d = 5 и 6), то величина
E
h
2
зависит от параметра X немонотонно. Аналогично ведут себя относительные флуктуации
E
S
2
(данные здесь не представлены), хотя их амплитуда значительно, примерно в 5 раз, меньше, чем амплитуда
E
h
2
. Последнее вполне объяснимо:
S
вычисляется с учетом взаимных положений всех атомов молекулы, тогда как h2 – только двух концевых атомов углерода. Отметим, что общие тенденции, которые отражают зависимости величины
E
h2 от параметра X (см. рис. 3), качественно согласуются с тенденциями изменения от этого параметра величины гибкости, характеризуемой отношением
О 4 8 12 16 4 8 12 16 4 8 12 16 8 12 8 12 12 16
X
Рис. 3. Зависимости относительных флуктуаций £ ^ 2 квадратов расстояний между концевыми атомами углерода невозмущенных углеводородных молекул от параметра X. Условия и обозначения, как на рис. 1
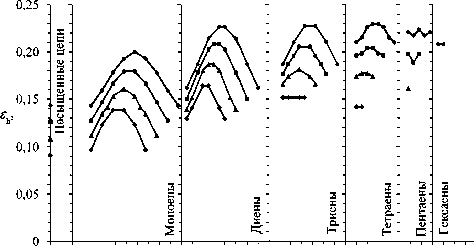
Удельная конформационная теплоемкость . Конформационная теплоемкость, приходящаяся на один угол вращения вокруг простой связи C-C, соседней с двойной (в ненасыщенных цепях), меньше, чем вокруг простой связи C-C в насыщенном участке цепи. Этот вывод следует из сравнения данных, например, для цепей 16:0 и 22:6 w 3 cis (рис. 4). Для обеих молекул Nφ = 13. Величина CV/Nφ для цепи 16:0 приходит-сяφ на угол вращения толφько вокруг простой связи C-C (углов другого типа Nφ не содержит). Для цепи 22:6 w 3 cis она почти полностью приходится на угол вращения вокруг связи C-C, соседней с двойной (таких углов 12 из 13). Различие между величинами C /N вызвано тем, что, как
Vφ упоминалось выше, первый торсионный барьер (~3 ккал/моль) больше, чем второй (~2 ккал/моль); минимумы поверхностей энергии в ненасыщенной цепи более пологие. В результате плотность высокоэнергетических состояний в насыщенной цепи оказывается больше, чем в ненасыщенной. С увеличением в молекуле количества двойных связей, то есть с ростом доли простых C-C
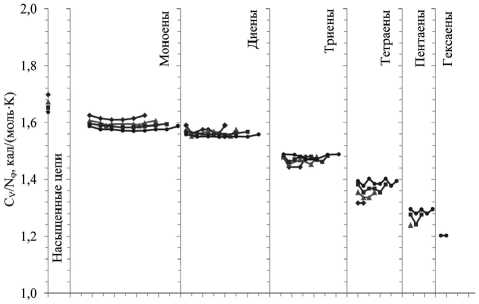
0 4 8 12 16 4 8 12 16 4 8 12 16 8 12 8 12 12 16 X
Рис. 4. Зависимости от параметра X удельных конформационных теплоемкостей C /N , связанных с флуктуаци-
Vφ ями энергии ближних взаимодействий невозмущенных углеводородных молекул. Условия и обозначения, как на рис. 1
связей, соседних с двойными, величина CV/Nφ молекулы постепенно уменьшается (см. рис. 4)φ. Удельная теплоемкость в цепях с N = Const, d = Const почти не зависит от X (см. рис. 4), поскольку при этом доля простых C-C связей, примыкающих к двойным, остается постоянной. Вклад внутреннего вращения в теплоемкость различных молекул цепного строения весьма важен; он обсуждается, например, в обзоре [21].
Итак, в работе установлена связь между химическим строением широко распространенных в природе цепных углеводородных молекул и относительными флуктуациями их геометрических размеров, а также конформационной теплоемкостью, связанной с флуктуациями энергии ближних взаимодействий, обобщен ряд тенденций изменения равновесной гибкости молекул. Полученные данные способствуют углублению общего понимания взаимосвязей между структурой и свойствами данного класса молекул.
-
* Работа выполнена при поддержке программ президента РФ «Ведущие научные школы» (гранты НШ-1642.2012.4, НШ-1410.2014.4).
Список литературы Изучение геометрических, термодинамических свойств и гибкости углеводородных молекул методом Монте-Карло
- Геннис Р. Биомембраны: Молекулярная структура и функции. М.: Мир, 1997. 624 с.
- Дашевский В. Г. Конформации органических молекул. М.: Химия, 1974. 428 с.
- Журкин Д. В., Рабинович А. Л. Оценка формы цепных углеводородных молекул методом Монте-Карло//Ученые записки Петрозаводского государственного университета. Сер. «Естественные и технические науки». 2014. № 6 (143). С. 109-117.
- Рабинович А. Л. Цепные молекулы как компоненты мембранных систем: компьютерное моделирование//Методы компьютерного моделирования для исследования полимеров и биополимеров. М.: Книжный дом «ЛИБРОКОМ», 2009. С. 410.
- Рабинович А. Л., Жу р к и н Д. В. Существенная выборка при моделировании непрерывного спектра конформаций макромолекул методом Монте-Карло//Труды Карельского научного центра Российской академии наук. Сер. «Математическое моделирование и информационные технологии». 2013. Вып. 4. С. 96-111.
- Флори П. Статистическая механика цепных молекул. М.: Мир, 1971. 440 с.
- Baschnagel J., Qin K., Paul W., Binder K. Monte Carlo Simulation of Models for Single Polyethylene Coils//Macromolecules. 1992. Vol. 25. № 12. P. 3117-3124.
- Bessières D., Pineiro M. M., De Ferron G., Plantier F. Analysis of the orientational order effect on n-alkanes: Evidences on experimental response functions and description using Monte Carlo molecular simulation//J. Chem. Phys. 2010. Vol. 133. 074507.
- Brown D., Clarke J. H. R., Okuda M., Yamazaki T. A molecular dynamics study of chain configurations in n-alkane-like liquids//J. Chem. Phys. 1994. Vol. 100. № 2. P. 1684-1692.
- Dettenmaier M. Conformation of n-alkane molecules in the melt and in cyclohexane solution studied by small-angle neutron scattering//J. Chem. Phys. 1978. Vol. 68. № 5. P. 2319-2322.
- Feller S. E., Gawrisch G., Mac K er ell Jr. A. D. Polyunsaturated Fatty Acids in Lipid Bilayers: Intrinsic and Environmental Contributions to Their Unique Physical Properties//J. Am. Chem. Soc. 2002. Vol. 124. № 2. P. 318-326.
- Ferguson A. L., Debenedetti P. G., Panagiotopoulos A. Z. Solubility and Molecular Conformations of n-Alkane Chains in Water//J. Phys. Chem. B. 2009. Vol. 113. № 18. P. 6405-6414.
- Goodsaid-Zalduondo F., Engelman D. M. Conformation of liquid n-alkanes//Biophys. J. 1981. Vol. 35. P. 587-594.
- Högberg C. J., Nikitin A. M., Lyubartsev A. P. Modification of the CHARMM Force Field for DMPC Lipid Bilayer//J. Comput. Chem. 2008. Vol. 29. P. 2359-2369.
- Mondello M., Gre s t G. S. Viscosity calculations of n-alkanes by equilibrium molecular dynamics//J. Chem. Phys. 1997. Vol. 106. № 22. P. 9327-9336.
- Nelson D. L., Cox M. M. Lehninger Principles of Biochemistry. 5th ed. N. Y.: Freeman W.H. and Co., 2008. Ch. 10. P. 343.
- Rabinovich A. L., R ip at t i P. O. Monte Carlo simulations of hydrocarbon oligomeric chains. Shape and dimension characteristics//Proc. SPIE. 2001. Vol. 4348. P. 225-236.
- Rabinovich A. L., R ip att i P. O. Monte Carlo simulations of hydrocarbon oligomeric chains: carbon skeleton cross sectional areas//Proc. SPIE. 2002. Vol. 4627. P118-128.
- Rey A., Kolinski A., Skolnick J. Effect of double bonds on the dynamics of hydrocarbon chains//J. Chem. Phys. 1992. Vol. 97. № 2. P. 1240-1249.
- Sun L., Siepmann J. I., Schure M. R. Conformation and Solvation Structure for an Isolated n-Octadecane Chain in Water, Methanol, and Their Mixtures//J. Phys. Chem. B. 2006. Vol. 110. P. 10519-10525.
- Wunderlich B. Phases of Amorphous, Crystalline, and Intermediate Order in Microphase and Nanophase Systems//Hot Topics in Thermal Analysis and Calorimetry. Vol. 8: Glassy, Amorphous and Nano-Crystalline Materials. Thermal Physics, Analysis, Structure and Properties. Dordrecht etc.: Springer Science+Business Media B.V., 2011. P. 93.
- Yoon D. Y., Flory P. J. Small angle neutron scattering by n-alkane chains//J. Chem. Phys. 1978. Vol. 69. № 6. P. 2536-2538. Zhurkin D. V., Petrozavodsk State University (Petrozavodsk, Russian Federation) Rabinovich A. L., Institute of Biology of Karelian Research Centre of RAS (Petrozavodsk, Russian Federation)
