Изучение структуры генофондной популяции русской белой породы кур методом геномного SNP-сканирования

Автор: Дементьева Н.В., Романов М.Н., Кудинов А.А., Митрофанова О.В., Станишевская О.И., Терлецкий В.П., Федорова Е.С., Никиткина Е.В., Племяшов К.В.
Журнал: Сельскохозяйственная биология @agrobiology
Рубрика: Генетическая структура популяций
Статья в выпуске: 6 т.52, 2017 года.
Бесплатный доступ
Популяция русских белых кур селекционировалась в генофондном хозяйстве Всероссийского НИИ генетики и разведения сельскохозяйственных животных (ВНИИГРЖ) в течение 25 поколений с использованием индивидуального подбора. Особенности этой породы - устойчивость к пониженной температуре выращивания в ранний постнатальный период и белый цвет эмбрионального пуха. В 2002-2012 годах ее разведение осуществлялось методом панмиксии, и к настоящему времени на основе сохранившегося поголовья сформирована новая популяция русских белых кур. Нашей целью было показать возможности полногеномного SNP-сканирования (single nucleotide polymorphisms) для изучения генетических особенностей структуры популяции малочисленных пород кур отечественного происхождения и динамические изменения молекулярной архитектуры на примере сравнения современной популяции русской белой породы с предковой популяцией 2001 года. Были проанализированы две группы кур: популяция 2001 года (6 гол., неродственные особи из двух линий) и современная популяция (156 гол.). SNP-анализ включал скрининг 162 образцов ДНК с помощью микрочипа Illumina Chicken 60K SNP iSelect BeadChip («Illumina», США). Контроль качества генотипирования, определение генетической структуры популяции методом многомерного шкалирования (multidimensional scaling, MDS), расчет показателей неравновесного сцепления (linkage disequilibrium, LD) и частоты встречаемости аллеей по группам проводили в программе PLINK 1.9. Построение модели разграничения кластеров на основе SNP-генотипов осуществляли с использованием программы ADMIXTURE. По результатам MDS-анализа современная популяция была условно разделена на четыре MDS-группы, что в сравнении с данными родословной адекватно отражает происхождение изученных особей. Представители предковой популяции были генетически сходны с группой MDS3. С применением F-статистики многофакторного дисперсионного анализа выявлено достоверное влияние группы, хромосомы, хромосомы в группе и дистанции между SNP-маркерами на значения LD (r2). В группе 2001 года по всем хромосомам наблюдались максимальные показатели r2 и высокая частота встречаемости LD, равного 1, при расстоянии между SNP-маркерами 500-1000 Кб. Количество мономорфных аллелей в этой группе также было самым высоким. На основании SNP-анализа сделан вывод о том, что современная популяция русских белых кур характеризуется распадом длинных LD-районов предковой популяции. Моделирование кластеров в программе ADMIXTURE выявило различия между группами современной популяции, определенными с помощью MDS-анализа. Группы, сформированные из особей, входящих в MDS1 и MDS2, имели однородную структуру и различались между собой при K = 4 и K = 5. Группа MDS4 образовывала генетически неоднородный кластер, отличающийся от групп MDS1 и MDS2 при значениях K от 2 до 5. Группа MDS3 была филогенетически близка к популяции 2001 года (при K от 2 до 5). Таким образом, анализ современной генофондной популяции русских белых кур показал ее неоднородность и сходство группы MDS3 с предковой популяцией 2001 года, которая, в свою очередь, характеризовалась большим числом мономорфных аллелей и высокой частотой встречаемости длинных LD-районов. SNP-сканирование позволило оценить генетическое сходство особей и популяционную структуру русской белой породы кур. Понимание генетической структуры важно при панмиктическом разведении и отслеживании исторических изменений в молекулярной организации генома генофондной популяции с ограниченным поголовьем.
Структура популяции, генетическое разнообразие, snp-сканирования, русская белая порода кур
Короткий адрес: https://sciup.org/142214095
IDR: 142214095 | УДК: 636.52/.58:575.174.5 | DOI: 10.15389/agrobiology.2017.6.1166rus
Studying the structure of a gene pool population of the Russian white chicken breed by genome-wide SNP scan
Period and white colour of the embryonic down. In 2002-2012, breeding was carried out by panmixia, and by now a new population of the Russian White chickens has been formed on the basis of the surviving stock. Comparison of the genetic variability of this population and the archival DNA of representatives of the 2001 population using microarray screening technology will help to assess the population structure and the preservation of the unique characteristics of its genome. The material for the study was DNA extracted from 162 chicken blood samples. Two groups of the Russian White breed were studied, the 2001 population and the current population. Genome-wide analysis using single nucleotide markers (SNP) included screening by means of the Illumina Chicken 60K SNP iSelect BeadChip microarray. Quality control of genotyping, determination of the population genetic structure by multidimensional scaling (MDS), calculation of linkage disequilibrium (LD) and allele frequency in the groups were carried out using PLINK 1.9 software program. The construction of a cluster delimitation model based on SNP genotypes was carried out using the ADMIXTURE program. According to the MDS analysis results, the current population can be divided into four MDS groups, which, when compared to the data of the pedigree, adequately reflect the origin of the studied individuals. The representatives of the ancestral population were genetically similar to the MDS3 group of the current population. Using the F-statistic of the two-way analysis of variance, a significant effect of the group, chromosome, chromosome in the group, and the distance between SNP markers on LD (r2) values was observed. In the 2001 group, the maximum r2 and the high incidence of LD equal to 1 were observed for all chromosomes, with a distance between SNP markers being 500-1000 Kb. There was also the greatest number of monomorphic alleles in this group. Based on the SNP analysis, we may conclude that the current Russian White chicken population is characterized by the disintegration of long LD regions of the ancestral population. Modelling clusters using the ADMIXTURE program revealed differences between the current population groups determined by MDS analysis. The groups composed of individuals included in MDS1 and MDS2 had a homogeneous structure and differed from each other at K = 4 and K = 5. The MDS4 group formed a genetically heterogeneous cluster different from the MDS1 and MDS2 groups at K of 2-5. The MDS3 group was phylogenetically close to the 2001 population (at K of 2-5). In general, the analysis of the current gene pool population of the Russian White chickens showed its heterogeneity while one of its groups (MDS3) was similar to the ancestral population of 2001, which in turn is characterized by a large number of monomorphic alleles and a high frequency of long LD regions. Thus, SNP scanning allowed evaluating the genetic similarity of individuals and the population structure of the Russian White chicken breed. Understanding the genetic structure is an important point in the panmictic breeding and tracking of historical changes in the molecular organization of the genome of a gene pool population with a limited number of animals.
Текст научной статьи Изучение структуры генофондной популяции русской белой породы кур методом геномного SNP-сканирования
Современные методы не находят заметного применения при углубленном изучении отечественного генофонда домашних кур. Вместе с тем сохранение и использование полезных качеств их непромышленных пород остаются важной научной и экономической задачей. Генофондная птица может использоваться в биотехнологии, служить моделью для изучения
* Исследования проведены при поддержке Российского научного фонда (грант ¹ 16-16-04060). Образцы крови получены из «Генетической коллекции редких и исчезающих пород кур» ВНИИРГЖ в рамках программы Федерального агентства научных организаций по поддержке биоресурсных коллекций.
биологических процессов и поиска ассоциаций генов (генетических маркеров) с хозяйственно полезными признаками (1-4).
Порода русских белых кур яичного направления продуктивности с белой окраской оперения разводится в генофондном хозяйстве Всероссийского НИИ генетики и разведения сельскохозяйственных животных (ВНИ-ИГРЖ) с 1953 года и изначально имела линейную структуру (1). Две линии (¹ 10 и ¹ 16) различались между собой по адаптационным способностям в условиях пониженной температуры выращивания в ранний постнатальный период (1). Еще одна опытная группа этой породы характеризовалась белым цветом эмбрионального пуха, и вся популяция на протяжении 25 поколений выращивалась при пониженных температурах (1). При разведении русских белых кур применяли индивидуальный подбор. До 2002 года популяция воспроизводилась внутри линий ¹ 10 и ¹ 16, а затем до 2012 года ее содержали при общепринятой температуре и разводили методом панмиксии, в результате чего линейная структура породы была утрачена. На основе сохранившегося поголовья была сформирована новая популяция русских белых кур, особенности которой — белый цвет пуха в 1-суточном возрасте и способность цыплят в первые дни жизни адаптироваться к пониженным температурам (22-23 ° С) в сравнении с общепринятыми в этом возрасте (30-33 ° С). В настоящее время содержание при пониженных температурах не применяется (1).
Совершенствование желательных качеств при разведении малочисленных популяций невозможно без оценки их популяционно-генетической структуры. Применение молекулярных маркеров (минисателлиты, микросателлиты) и другие методы изучения полиморфизма ДНК, широко использовавшиеся ранее (2-7), в последнее время уступают место использованию многочисленных однонуклеотидных маркеров (single nucleotide polymorphisms, SNP). Десятки и сотни тысяч SNP-маркеров позволяют генотипировать весь геном и связывать найденные вариации по этим маркерам с количественными признаками. SNP-сканирование — высокоэффективный инструмент генетического анализа, способный выявлять структурные особенности популяции, которые можно использовать в селекции (8-11). Сочетание молекулярно-генетических данных с математическими моделями повышает точность прогноза племенной ценности животных для селекции и эффективности управления генетической изменчивостью, что приводит к ускорению генетического прогресса в селекционируемых популяциях (12, 13). При разведении малочисленных популяций наблюдается рост частоты встречаемости протяженных участков гаплотипов (в том числе ROH, гомозиготных районов), достаточно стабильно передающихся из поколения в поколение (14-16), что ведет к снижению генетического разнообразия в малой популяции (17, 18).
Для оценки дифференциации изучаемых групп (популяций, пород) широко используется многомерное шкалирование (multidimensional scaling, MDS-анализ) (19). Разработаны методы, в основу которых положены предопределенная структура анализируемых групп и расчет генетического расстояния между особями с помощью алгоритма построения дерева, объединяющего их в кластеры (20). Созданы и применяются модели для разграничения кластеров на основе генотипов по десяткам тысяч локусов и с использованием байесовского подхода, которые, например в компьютерных программах STRUCTURE и ADMIXTURE, учитывают также наличие распределения Харди-Вайнберга и неравновесного сцепления (linkage disequilibrium, LD) (7, 21, 22).
С помощью полногеномного SNP-генотипирования нами впервые выявлена субпопуляционная структура современной породы кур русская 1167
белая из биоресурсной коллекции ВНИИРГЖ, которая располагает уникальным генетическим материалом отечественных и зарубежных пород, и установлены отличия изученных групп птицы от исходной популяции.
Нашей целью было показать возможности полногеномного SNP-сканирования для изучения генетических особенностей структуры популяции малочисленных пород кур отечественного происхождения и динамические изменения молекулярной архитектуры на примере сравнения современной популяции русской белой породы с популяцией 2001 года.
Методика. Материалом для исследования служила ДНК, выделенная из крови кур ( Gallus gallus ) русской белой породы, содержащихся в биоре-сурсной коллекции ВНИИРГЖ «Генетическая коллекция редких и исчезающих пород кур» (г. Санкт-Петербург—Пушкин). Были проанализированы две группы кур: популяция 2001 года (6 гол., неродственные особи из двух линий) и современная популяция (156 гол.). SNP-анализ включал скрининг 162 образцов ДНК с микрочипом Illumina Chicken 60K SNP iSelect BeadChip («Illumina», США). Качество генотипированных SNP-локусов контролировали с помощью программы PLINK 1.9 (23). Дополнительно для анализа отбирали образцы ДНК с качеством генотипирования по SNP-локусам более 90 %, что оценивалось с использованием программы GenomeStudio («Il-lumina», США). Были установлены лимиты по ошибкам Харди-Вайнберга — HWE (P ≤ 0,0001). SNP, находящиеся в неравновесном сцеплении (--indeppairwise 50 5 0,5) в программе PLINK 1.9, удаляли. Для устранения влияния пола на оценку были исключены SNP-маркеры, находящиеся на половых хромосомах. Генетическую структуру популяции выявляли с помощью MDS-анализа в программе PLINK 1.9. Частоту встречаемости аллеей и неравновесное сцепление по группам также рассчитывали с помощью PLINK 1.9.
Многофакторный дисперсионный анализ (ANOVA) проводили в программе RStudio (24). Достоверность влияния группы, хромосомы, их взаимодействия и SNP-интервала (п.н.) на LD оценивали с помощью линейной модели (10): r2 i j = μ + BL i + Gga j + (BL ½ Gga) i j + bSNP int + e ik , где r2 i j — парное значение LD, μ — общее среднее значение LD, BL i — эффект i -й группы, Gga j — эффект j -й хромосомы курицы (с 1-й по 28-ю хромосому), SNPint — эффект интервала между SNP-маркерами, который определялся как расстояние между маркерами (число пар нуклеотидов), b — коэффициент регрессии.
Построение модели для разграничения кластеров на основе SNP-генотипов проводили в программе ADMIXTURE (25).
Результаты. В зависимости от распределения генотипированных особей популяцию условно разделили на MDS-группы и сравнивали с данными родословной и с представителями предковой популяции 2001 года. На плотность распределения точек на графике, полученном методом многомерного шкалирования (рис. 1), оказывало влияние наличие мономорфных и минорных аллелей с низкой частотой (MAF). Это усложняло оценку изменчивости по остальным маркерам. В предварительной работе по подбору параметров фильтрации MAF было выбрано ограничение 0,1, исключающее все мономорфные аллели и минорные с частотой ниже 10 %.
Условно всю современную популяцию можно было разделить на четыре кластера (MDS1-MDS4). Кластеры MDS1 и MDS2 оказались разделены по оси C2, MDS1, MDS2, MDS3 от MDS4 — по оси C1. В кластер MDS1 входили в основном особи, ведущие происхождение от петуха ¹ 99. Кластер MDS2 представляли потомки петухов ¹ 98 и ¹ 99 (табл. 1). Кластер MDS4 преимущественно составляла птица, родоначальником которой был петух ¹ 97. Кластер MDS3 объединял потомков петуха ¹ 58 и промежуточные варианты, близкие по происхождению матерей к другим кластерам.
|
1. Распределение потомков петухов по MDS-клас- Из выделенных терам в современной популяции кур русской белой кластеров были сфор- породы (биоресурсная коллекция ВНИИРГЖ мированы группы осо- «Генетическая коллекция редких и исчезающих бей, у которых изучали пород кур», г. Санкт-Петербург—Пушкин) генетические характе- |
||
|
Петух- Петух-родо-отец, ¹ начальник, ¹ |
Кластер |
ристики (табл. 2). Стру- Всего ктурные особенности |
|
MDS1 MDS2 MDS3 MDS4 |
||
|
981206 98 0 10 3 0 13 групп оценивали по на- 981205 98 0 16 0 1 17 981501 98 0 0 5 0 5 личию и протяженно- 991803 99 0 14 0 0 14 сти районов, где на- 991203 99 16 0 1 0 17 970905 97 0 0 0 12 12 блюдалось неравновес- 971103 97 0 0 0 16 16 ное сцепление SNP-ма- 970907 97 0 0 0 15 15 971601 97 0 0 0 13 13 ркеров. Максимальное 581706 58 0 0 9 0 9 среднее значение LD 481701 58 1 0 8 7 16 было обнаружено в по- 639 0 0 9 0 9 Всего 17 40 35 64 156 пуляции 2001 года. Ко личество мономорфных |
||
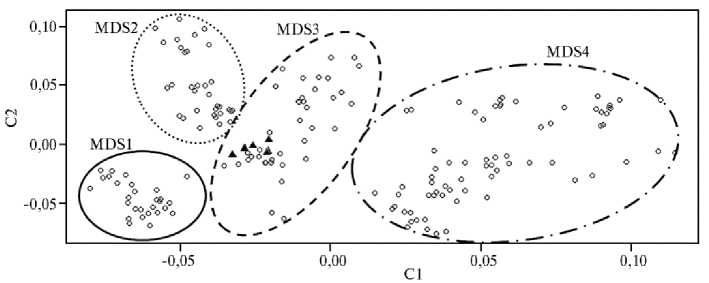
Рис. 1. MDS-распределение генотипированных кур русской белой породы по четырем кластерам (MDS1-MDS4): о — особи современной популяции, ▲ — архивные образцы ДНК 2001 года; С1 — координата 1, С2 — координата 2 (б иоресурсная коллекция ВНИИРГЖ «Генетическая коллекция редких и исчезающих пород кур», г. Санкт-Петербург—Пушкин).
аллелей в этой группе также оказалось самым высоким. Группа MDS3 занимала центральную позицию вместе с популяцией 2001 года, но в отличие от нее имела минимальное число мономорфных SNP и существенное количество минорных аллелей с частотой встречаемости меньше 10 %. В группе 2001 года по всем хромосомам наблюдались максимальные показатели r2 и высокая частота (0,24) неравновесного сцепления, равного 1, при значительном расстоянии между SNP-маркерами (500-1000 т.п.н.). Среднее значение LD, рассчитанное по хромосомам, было высоким как в современной, так и в предковой популяции и колебалось от 0,150±0,006 до 0,587±0,006.
Дополнительный многофакторный дисперсионный анализ с использованием F-теста (табл. 3) показал достоверное (Р < 0,0001) влияние на LD (r2) группы, хромосомы, дистанции между SNP-маркерами и хромосомы в группе. Наибольшее влияние оказывала группа и дистанция между SNP-маркерами.
Одним из эффективных методов выявления различий между группами и породами животных считается кластерный анализ адмикс-моделей (25, 26). При кластеризации в программе ADMIXTURE была проведена оценка коэффициента кросс-проверки (CV) для определения оптимального числа K. Минимальная ошибка наблюдалась при K = 12 (рис. 2). Группы MDS1 и MDS2 имели однородную структуру и не различались при К = 2, частично различались при К = 3 и обнаруживали значительное различие при
К = 4 и К = 5. Группа MDS4 образовала генетически неоднородный кластер, отличающийся от MDS1 и MDS2 при К от 2 до 5. Группа MDS3 была более однородна и близка к популяции 2001 года при величине К от 2 до 5.
2. Характеристика предковой популяции (2001 год) и групп, сформированных на основе MDS-кластеров распределения особей современной генофондной популяции кур русской белой породы, по локусам SNP-маркеров (биоре-сурсная коллекция ВНИИРГЖ «Генетическая коллекция редких и исчезающих пород кур», г. Санкт-Петербург—Пушкин)
|
Показатель |
Группа |
||||
|
MDS1 |
MDS2 |
MDS3 |
MDS4 |
2001 |
|
|
Всего генотипировано SNP В том числе: |
57636 |
57636 |
57636 |
57636 |
57636 |
|
локусов с высоким качеством генотипирования (> 0,90) |
43224 |
43224 |
43224 |
43224 |
43224 |
|
локусов с мономорфными аллелями |
9176 |
8157 |
1507 |
5393 |
19833 |
|
локусов с минорными аллелями (MAF ≤ 0,1) |
5943 |
7800 |
10443 |
8200 |
3827 |
|
HWE (P ≤ 0,0001) |
949 |
1021 |
1244 |
1543 |
0 |
|
LD ( M ±SEM) Частота LD = 1 при расстоянии между |
0,272±0,001 |
0,241±0,001 |
0,193±0,001 |
0,197±0,001 |
0,506±0,001 |
|
SNP 500-1000 Кб |
0,07 |
0,03 |
0,02 |
0,02 |
0,24 |
П р и м еч а ни е. MAF — частота минорных аллелей, HWE — число SNP, не прошедших тест на равновесие Харди-Вайнберга (при P ≤ 0,0001), LD — неравновесное сцепление, M — среднее значение LD, ±SEM — стандартная ошибка среднего.
3. Оценка эффекта MDS-группы, хромосомы и интервала между SNP-маркерами на неравновесие по сцеплению (r2) в популяции кур русской белой породы (биоресурсная коллекция ВНИИРГЖ «Генетическая коллекция редких и исчезающих пород кур», г. Санкт-Петербург—Пушкин)
|
Фактор |
п DF |
] SS |
1 MS |
] F |
1 P |
|
Группа |
4 |
3244 |
811,0 |
11947,40 |
P < 0,001 |
|
Хромосома |
27 |
178 |
6,6 |
96,93 |
P < 0,001 |
|
SNP-дистанция |
1 |
244 |
243,8 |
3592,35 |
P < 0,001 |
|
Группа ½ хромосома |
105 |
289 |
2,8 |
40,61 |
P < 0,001 |
|
Примечание. DF — |
число степеней свободы, SS — |
сумма квадратов, |
MS — средняя |
квадратов, F — |
|
|
распределение Фишера. |
|||||
Как известно, традиционный подход в селекции птицы при индивидуальном учете включает отбор по продуктивности родителей, собственной продуктивности и подбор неродственных пар. При панмиктическом разведении, имевшем место в случае современной популяции русских белых кур, сложно определить происхождение и генетическую изменчивость полученного потомства. Характеристика генетической изменчивости решает проблему определения структуры и оценивает динамические изменения молекулярной архитектуры в популяции.
В современной популяции русских белых кур как потомков популяции 2001 года мог происходить специфический дрейф генов. Отметим, что представители популяции 2001 года (2 особи линии ¹ 16 и 4 особи линии ¹ 10) локализовались в центральной области распределения одного и того же современного кластера MDS3 (см. рис. 1). Хотя число обследованных особей предковой популяции было невелико, данные их генотипирования можно принимать в расчет, поскольку значительное число SNP-маркеров, использованных в популяционном анализе, не требует для объективной оценки увеличения числа анализируемых особей (27-29). Неродственные особи разных линий были расположены очень близко друг от друга, так как обе линии вели происхождение от одного петуха.
Однако генотипы неродственных особей, участвовавших в анализе, видимо, не полностью отражают генетическое разнообразие породы в 2001 году. Не все минорные аллели популяции 2001 года при ограниченном наличии материала (6 гол.) оказались учтены. Возможно, некоторые мономорф-1170
ные аллели в популяции были минорными. В то же время при MDS-анализе минорные аллели (частично) и все мономорфные элиминировались при фильтрации MAF и не могли повлиять на распределение и, соответственно, выводы о генетической близости или удаленности особей и популяций. В предварительном анализе полное исключение минорных аллелей не влияло на картину разделения популяции на MDS-группы. Удаление SNP, находящихся в неравновесном сцеплении (--indep-pairwise 50 5 0,5), при варьировании условий отбора SNP не изменило расположения кластеров. Результаты наших исследований показали, что общность происхождения служит основным фактором, влияющим на относительное MDS-распределение.
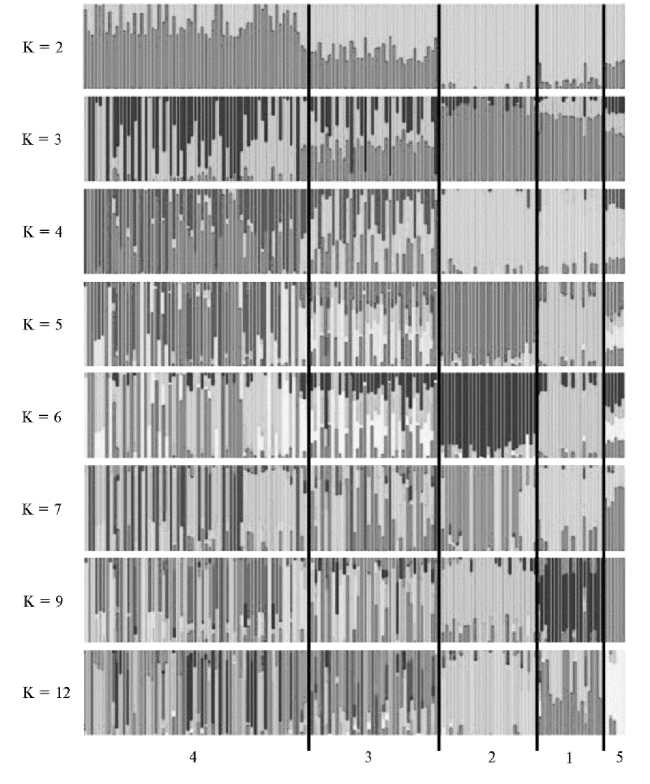
Рис. 2. Популяционная изменчивость SNP-маркеров внутри русской белой породы кур, рассчитанная в программе ADMIXTURE: 1, 2, 3, 4 — кластеры MDS1, MDS2, MDS3 и MDS4 современной популяции, 5 — представители популяции 2001 года. К — выбранное число предковых популяций (биоресурсная коллекция ВНИИРГЖ «Генетическая коллекция редких и исчезающих пород кур», г. Санкт-Петербург—Пушкин).
Наличие уникальных гаплотипов — важная характеристика популяций (19, 29). Протяженность районов, где наблюдается неравновесное сцепление SNP-маркеров, считается основной структурной особенностью исследуемых групп (8). Большое количество маркеров, находящихся в неравновесном сцеплении, служило отличительной чертой предковой популяции русской белой породы, что отражалось на величине LD. Наличие у неродственных животных значительного количества SNP, находящихся в неравно- весном сцеплении на большом расстоянии друг от друга, свидетельствует скорее об ограниченном поголовье, которое было использовано в селекции. Это подтверждают другие исследования на промышленных линиях (8, 17). Современная популяция русских белых кур характеризуется распадом длинных LD районов и сокращением частоты гаплотипов предковой популяции.
Таким образом, неоднородность современной популяции русских белых кур из генофондного хозяйства Всероссийского НИИ генетики и разведения сельскохозяйственных животных обусловлена их происхождением от разных петухов-родоначальников. Обнаружена группа (MDS3), имеющая наибольшее сходство с предковой популяцией 2001 года. Особенность последней — значительное число мономорфных аллелей и высокая частота встречаемости длинных LD-районов. В современной популяции увеличилась частота минорных аллелей и уменьшились показатели LD. В целом SNP-сканирование позволяет выявлять структуру родственных связей в породе на основе генетического сходства индивидуумов, что особенно важно при панмиктическом разведении пород с ограниченным поголовьем. Сравнение современной популяции с предковой дает возможность проследить исторические изменения в молекулярной организации генома русской белой породы. Генофондные популяции, генетическая изменчивость которых формировалась в течение длительного времени, считаются ценным источником биоразнообразия. Характеристика их генетических особенностей актуальна для использования лучших качеств животных в селекции по хозяйственно полезным признакам. В настоящей работе получена важная информация о генетических особенностях малочисленной породы кур отечественного происхождения. Эти сведения в дальнейшем могут применяться для управления, сохранения и использования ценных генетических ресурсов, а также отслеживания динамических изменений в молекулярной организации генома генофондной популяции с ограниченным поголовьем.
Список литературы Изучение структуры генофондной популяции русской белой породы кур методом геномного SNP-сканирования
- Соколова А.Н. Генетико-селекционные методы создания популяции кур с повышенной устойчивостью к неоплазмам. Автореф. докт. дис. СПб-Пушкин, 1999.
- Weigend S., Romanov M.N. Current strategies for the assessment and evaluation of genetic diversity in chicken resources. World Poultry Sci. J., 2001, 57(3): 275-288 ( ) DOI: 10.1079/WPS20010020
- Soller M., Weigend S., Romanov M.N., Dekkers J.C.M., Lamont S.J. Strategies to assess structural variation in the chicken genome and its associations with biodiversity and biological performance. Poultry Sci., 2006, 85(12): 2061-2078 ( ) DOI: 10.1093/ps/85.12.2061
- Romanov M.N., Weigend S. Analysis of genetic relationships between various populations of domestic and jungle fowl using microsatellite markers. Poultry Sci., 2001, 80(8): 1057-1063 ( ) DOI: 10.1093/ps/80.8.1057
- Dunn I.C., Miao Y.-W., Morris A., Romanov M.N., Wilson P.W., Waddington D. A study of association between genetic markers in candidate genes and reproductive traits in one generation of a commercial broiler breeder hen population. Heredity, 2004, 92(2): 128-134 ( ) DOI: 10.1038/sj.hdy.6800396
- Тыщенко В.И., Митрофанова О.В., Дементьева Н.В., Терлецкий В.П. Оценка генетического разнообразия в породах и экспериментальных популяциях кур с помощью ДНК-фингерпринтинга. Сельскохозяйственная биология, 2007, 4: 29-33.
- Mekchay S., Supakankul P., Assawamakin A., Wilantho A., Chareanchim W., Tongsima S. Population structure of four Thai indigenous chicken breeds. BMC Genet., 2014, 15: 40 ( ) DOI: 10.1186/1471-2156-15-40
- Qanbari S., Hansen M., Weigend S., Preisinger R., Simianer H. Linkage disequilibrium reveals different demographic history in egg laying chickens. BMC Genet., 2010, 11: 103 (doi 10.1186/1471-2156-11-103).
- Яковлев А.Ф., Смарагдов М.Г. Значительное повышение точности оценки племенной ценности животных в молочном скотоводстве. Зоотехния, 2011, 5: 2-4.
- Khanyile K.S., Dzomba E.F, Muchadeyi F.C. Population genetic structure, linkage disequilibrium and effective population size of conserved and extensively raised village chicken populations of Southern Africa. Front. Genet., 2015, 6: 13 ( ) DOI: 10.3389/fgene.2015.00013
- Smaragdov M.G., Saksa E.I., Kudinov A.A., Dementeva N.V., Mitrofanova O.V., Plemyashov K.V. Genome-wide analysis of across herd Fst heterogeneity in holsteinized cattle. Russ. J. Genet., 2016, 52(2): 173-179 ( ) DOI: 10.1134/S1022795416020150
- Habier D., Fernando R.L., Dekkers J.C.M. The impact of genetic relationship information on genome-assisted breeding values. Genetics, 2007, 177(4): 2389-2397 ( ) DOI: 10.1534/genetics.107.081190
- Weng Z., Wolc A., Shen X., Fernando R.L., Dekkers J.C., Arango J., Settar P., Fulton J.E., O'Sullivan N.P., Garrick D.J. Effects of number of training generations on genomic prediction for various traits in a layer chicken population. Genet. Sel. Evol., 2016, 48: 22 ( ) DOI: 10.1186/s12711-016-0198-9
- Meuwissen T.H., Hayes B.J., Goddard M.E. Prediction of total genetic value using genome-wide dense marker maps. Genetics, 2001, 157(4): 1819-1829.
- Solberg T.R., Sonesson A.K., Woolliams J.A., Odegard J., Meuwissen T.H. Persistence of accuracy of genome-wide breeding values over generations when including a polygenic effect. Genet. Sel. Evol., 2009, 41: 53 ( ) DOI: 10.1186/1297-9686-41-53
- Fariello M.I., Boitard S., Naya H., SanCristobal M., Servin B. Detecting signatures of selection through haplotype differentiation among hierarchically structured populations. Genetics, 2013, 193(3): 929-941 ( ) DOI: 10.1534/genetics.112.147231
- Muir W.M., Wong G.K.-S., Zhang Y., Wang J., Groenen M.A.M., Crooijmans R.P.M.A., Megens H.-J., Zhang H., Okimoto R., Vereijken A., Jungerius A., Albers G.A.A., Lawley C.T., Delany M.E., MacEachern S., Cheng H.H. Genome-wide assessment of worldwide chicken SNP genetic diversity indicates significant absence of rare alleles in commercial breeds. PNAS, 2008, 105(45): 17312-17317 ( ) DOI: 10.1073/pnas.0806569105
- Fragomeni B.D.O., Misztal I., Lourenco D.L., Aguilar I., Okimoto R., Muir W.M. Changes in variance explained by top SNP windows over generations for three traits in broiler chicken. Front. Genet., 2014, 5: 332 ( ) DOI: 10.3389/fgene.2014.00332
- Beynon S.E., Slavov G.T., Farré M., Sunduimijid B., Waddams K., Davies B., Haresign W., Kijas J., MacLeod I.M., Newbold C.J., Davies L., Larkin D.M. Population structure and history of the Welsh sheep breeds determined by whole genome genotyping. BMC Genet., 2015, 16: 65 ( ) DOI: 10.1186/s12863-015-0216-x
- Saitou N., Nei M. The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol. Biol. Evol., 1987, 4(4): 406-425.
- Pritchard J.K., Stephens P., Donnelly P. Inference of population structure using multilocus genotype data. Genetics, 2000, 155(2): 945-959.
- Evanno G., Regnaut S., Goudet J. Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Mol. Ecol., 2005, 14(8): 2611-2620 ( ) DOI: 10.1111/j.1365-294X.2005.02553.x
- Chang C.C., Chow C.C., Tellier L.C., Vattikuti S.S., Purcell S.M., Lee J.J. Second-generation PLINK: rising to the challenge of larger and richer datasets. GigaScience, 2015, 4: 7 ( ) DOI: 10.1186/s13742-015-0047-8
- RStudio. Режим доступа: http://www.rstudio.com. Без даты.
- Alexander D.H., Novembre J., Lange K. Fast model-based estimation of ancestry in unrelated individuals. Genome Res., 2009, 19(9): 1655-1664 ( ) DOI: 10.1101/gr.094052.109
- Денискова Т.Е., Доцев А.В., Багиров В.А., Виммерс К., Рейер Х., Брем Г., Зиновьева Н.А. Оценка биоразнообразия у межвидовых гибридов рода Ovis с использованием STR-и SNP-маркеров. Сельскохозяйственная биология, 2017, 52(2): 251-260 ( ) DOI: 10.15389/agrobiology.2017.2.251rus
- Wragg D., Mwacharo J., Alcalde J., Hocking P.M., Hanotte O. Analysis of genome-wide structure, diversity and fine mapping of Mendelian traits in traditional and village chickens. Heredity, 2012, 109(1): 6-18 ( ) DOI: 10.1038/hdy.2012.9
- Ardlie K.G., Kruglyak L., Seielstad M. Patterns of linkage disequilibrium in the human genome. Nat. Rev. Genet., 2002, 3(4): 299-309 ( ) DOI: 10.1038/nrg777
- Andreescu C., Avendano S., Brown S. R., Hassen A., Lamont S.J., Dekkers J.C. Linkage disequilibrium in related breeding lines of chickens. Genetics, 2007, 177(4): 2161-2169 ( ) DOI: 10.1534/genetics.107.082206
