Кинетическая модель деактивации О2 ( а 1D)
")
Автор: Торбин Алексей Петрович, Першин Андрей Александрович, Азязов Валерий Николаевич
Журнал: Известия Самарского научного центра Российской академии наук @izvestiya-ssc
Рубрика: Физика и электроника
Статья в выпуске: 2-1 т.17, 2015 года.
Бесплатный доступ
Для повышения производительности электроразрядного кислородно-йодного лазера необходимо увеличивать давление О 2 на выходе электроразрядного генератора синглетного кислорода O 2( a 1D). Механизм деактивации O 2( a 1D) при повышенных давлениях кислорода до конца не изучен. В данной работе показано, что колебательно-возбужденный озон O 3( υ ), образованный в трехчастичном процессе рекомбинации O + O 2 + M ® O 3( υ ) + M, играет важную роль в деактивации синглетного кислорода в смесях O-O 2-O 3. В послеразрядной зоне главным каналом деактивации O 2( a 1D) является процесс O 3( υ ³2) + O 2( a 1D) ® 2O 2 + O. Если не принимать никаких мер по снижению концентрации атомов кислорода, то вклад этого процесса в общую скорость удаления синглетного кислорода будет значительным даже в разрядной зоне.
Синглетный кислород, атомы кислорода, лазерный фотолиз, деактивация, колебательно-возбужденный озон
Короткий адрес: https://sciup.org/148203601
IDR: 148203601 | УДК: 621.373.826
Kinetics deactivation model of О2 ( а 1D)
To improve the performance of electric oxygen-iodine laser it is necessary to increase the pressure of the O 2 in the output of electric singlet oxygen O 2( a 1D) generator. The mechanism of O 2( a 1D) deactivation under high oxygen pressures is not entirely clear. In this paper it is shown that vibrationally-excited ozone O 3( υ ), formed in the three-body recombination process O + O 2 + M ® O 3( υ ) + M, plays an important role in deactivation of singlet oxygen in O-O 2-O 3 gas mixtures. The main channel of O 2( a 1D) deactivation in post-discharge zone is process O 3( υ ³2) + O 2( a 1D) ® 2O 2 + O. If no action is taken to reduce the concentration of oxygen atoms, the contribution of this process to the overall removal rate of singlet oxygen will be significant even in the discharge zone.
Текст научной статьи Кинетическая модель деактивации О2 ( а 1D)
Низкий коэффициент усиления активной среды электроразрядного кислородно-йодного лазера (ЭКИЛ) [1] не позволяет осуществлять эффективный съем запасенной в синглетном кислороде О2( а 1∆) энергии [2]. Для повышения коэффициента усиления необходимо увеличивать концентрации как атомов йода, так и молекул О2( а 1∆). Обнаружено [3], что скорость деактивации О2( а 1∆) на выходе электроразрядного генератора синглетного кислорода (ЭГСК) растет с увеличением концентраций атомов кислорода [О], молекул кислорода [О2], а также буферного газа [M]. Темп убыли О2( а 1∆) на выходе ЭГСК удовлетворительно объясняется феноменологическим трехчастичным механизмом деактивации [3]
О + О2( а 1∆) + М → О + О2+ М. (1)
В [4-6] также наблюдали аномально высокую скорость деактивации О2( а 1∆) в смесях О-О2-Ar-He-CO2 в послефотолизной зоне. При этом добавление Ar не сказывалось на скорости деактивации, а добавление He и CO2 даже уменьшало темп деактивации О2( а 1∆) [6], что находится в противоречии с механизмом (1). Темп убыли O2( a 1D) в послефотолизной зоне хорошо объясняется химическим процессом [5, 6]
O2( a 1D) + O3( υ ) → 2O2+ O, (2)
где колебательно-возбужденная молекула озона O3( υ ) образуется в процессе трехчастичной рекомбинации [7]
О + О2+ М → О3( υ ) + М. (3)
Здесь υ = υ 1 + υ 2 + υ 3 – суммарное число квантов на деформационной υ 2 =701 см-1 , симметричной υ 1 =1103 см-1 и антисимметричной υ 3 =1042 см-1 валентных модах молекулы озона [7]. В ряде работ [7-10] также отмечается, что О3( υ ) эффективно реагирует с O2( a 1D). Несмотря на это, процесс (2) ранее не привлекался для объяснения высокого темпа убыли O2( a 1 Δ ) на выходе ЭГСК. Целью данной работы является определение влияния процесса (2) на динамику O2( a 1 Δ ) в ЭКИЛ.
РЕЗУЛЬТАТЫ И АНАЛИЗ
Кинетическая схема процессов с участием колебательно-возбужденного озона приведена в [6]. Отсутствие измеренных значений вероятностей возбуждения колебательных мод молекулы О3 в процессе (3) и констант скоростей процесса (2) для конкретных наборов значений υ 1 , υ 2 и υ 3 осложняет моделирование колебательной кинетики озона. В [5] показано, что упрощенная модель колебательной кинетики озона с объединенной модой υ , адекватно описывает экспериментальные результаты, полученные в [4-6]. В процессе (3) образуется молекула озона с υ ≥2 [5, 7]. В столкновениях с частицами среды колебательные кванты перераспределяются между тремя модами
O3( υ 1, υ 2, υ 3) + M ↔ O3( υ 1+1, υ 2, υ 3-1) + M
O3( υ 1, υ 2, υ 3) + M ↔ O3( υ 1, υ 2+1, υ 3-1) + M.
O3( υ 1, υ 2, υ 3) + M ↔ O3( υ 1-1, υ 2+1, υ 3) + M
O3( υ ) деактивируется в VT процессе [7]
О3( υ ) + М → О3( υ -1) + М, (4)
или удаляется в химических реакциях (2) и
O3( υ ) + О → O2 + O2. (5)
Скорости реакций (2) и (5) с υ <2 медленные и не оказывают существенного влияния на кинети-
Известия Самарского научного центра Российской академии наук, т.17, №2, 2015
ку O3( u ) [5]. Для и > 2 в [5] приводятся следующие значения констант скоростей реакций: к 2 = 4,1 х 1011 см 3 с -1 и к 5 = 1,2 х 1011 см 3 с -1.
Квазистационарная концентрация O3( u > 2) может быть найдена из баланса его образования в процессе (3) и убыли в процессах (2), (4) и (5):
1 k зМ [О][О 2 ][М]
М
Рз Ф > 2)] =
k 2 [O 2 ( a )] + 1 k 4M [M] + k 5[O] M . (6)
Покажем, что процесс (2) обеспечивает такие же скорости убыли О2(а1∆) как и процесс (1) для условий экспериментов работы [3], где впервые был предложен трехчастичный процесс деактивации. Для этого рассмотрим отношение скоростей этих процессов с учетом (6):
Q = k 2 [O 3 ( u> 2)][O 2 ( a )] 2/1 1 k ,M [O][O 2 ( a )][M]
M k 2[O2]1 k 3M[M] M
1 k M I k 2 [O 2 ( a )] + 1 k M [M] + к 5[О] M V M
Для условий экспериментов [3]: Ar:O2=99:1, давление смеси 100 Торр, концентрация молекул синглетного кислорода [O2( а )]=1,5 х 1015 см3, [O]=2 х 1015 см3, температура смеси T =300 K при к 1A =0,62 х 10-32 см6 с-1 [3], к 1 O2=1 х 10-32 см6 с-1 [3], к 3O2=6 х 10-34 см6 с-1 [3], к 3Ar=0,62 х к 3 02 [3], к 4 O2=3 х 10 —14 см3с-1 [7], к 4 Ar=5,9 х 10-15 см3 с-1 [7] отношение Q 2/1 близко к единице. Следовательно, процесс (2) обеспечивает темпы убыли О2(а1∆) на выходе ЭГСК.
Скорость процесса (2) с учетом (6) может быть представлена в виде:
k 2 [O 2 ( a )] 1 k 3M [O][O 2 ][M]
R = k 2 [O3 ( u > 2)][O 2 ( a )] =----------------------
-
2 2L л Л J k 2[O2( a )] + 1 k 4 M [M] + k 5[O].
M
Для типичных условий в послеразрядной зоне ЭГСК [1]: [He]= 9*1017 см3, [02]= 2,5 х 1017 см3, [0]=5 х 1015 см3, [O2( а )]=3*1016 см3, T =550 K слагаемые в знаменателе удовлетворяют условию k 2 [O 2 ( a )] >> 1 k 4M[M] + k 5[O] . В этом случае M
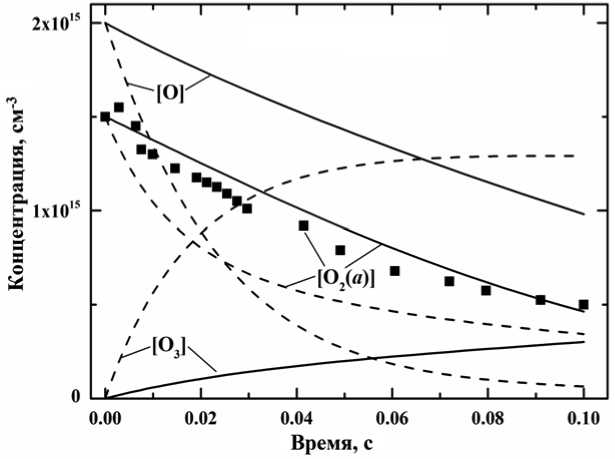
Рис. 1. Временные профили концентраций O, O2( а 1 Д ) и O 3 в послеразрядной зоне
R 2 « 1 k 3M [O][O2][M] и скорость процесса M
-
( 2) лимитируется скоростью образования O3( υ ) в процессе трехчастичной рекомбинации (3). Это явилось причиной, почему в [3] склонились в пользу трехчастичной деактивации O2( a 1 Δ ).
На рис. 1 представлены временные профили концентраций O, O2( a 1 Δ ) и O3 на выходе несамостоятельного разряда для условий эксперимента из [3]: состав газовой смеси Ar:O2 = 99:1, общее давление P = 100 Торр. Символами обозначены экспериментальные значения концентрации O2( a 1 Δ ) из [3]. Сплошными линиями на рисунке показаны расчетные значения концентраций согласно двухчастичному (процесс (2)), а пунктирными – трехчастичному (процесс (1)) механизму деактивации синглетного кислорода.
Из рис. 1 очевидно, что предложенная кинетическая модель двухчастичной деактивации синглетного кислорода обеспечивает лучшее согласование с экспериментально полученными значениями концентрации O2( a 1 Δ ), чем трехчастичная. Более того, предложенная модель предсказывает более низкие скорости удаления атомарного кислорода и наработки O3 вследствие регенерации атомов O в процессе (2). В результате согласно двухчастичному механизму деактивации O2( a 1 Δ ) концентрация атомов кислорода падает всего в два раза за 0,1 с , в то же время как в трехчастичной кинетической модели она снижается практически до нуля за такой же промежуток времени.
Реакция (2) может вносить заметный вклад в скорость убыли О2( а 1∆) также в разрядной зоне ЭГСК. Рассмотрим отношение скорости данной реакции к скорости самого быстрого процесса в разрядной зоне - деактивации О2( а 1∆) электронным ударом
О2( а 1 А) + е ^ О2 + е, (7) константа скорости которой к 7 = 1,02 х 10 -9 см 3 с -1[11].
Отношение скоростей процессов (2) и (7) определяется по формуле:
Q 2/7 = k JO,№ 2>][O ; ( a )] = k .- tO«Q .« Ml
k [e '°/ a >1 k j,/ k 7[O _( a >] , y k M[M] + k , [O]1 .
V M
Для экспериментальных условий [12] отношение скоростей процессов (2) и (7) составляет Q 2/7 « 0,2, в случае когда атомы О не удалялись из системы. В экспериментах с уменьшенной концентрации атомов О за счет добавления NO и покрытия стенок камеры окисью ртути отношение намного меньше Q 2/7 « 0,03. Следовательно, процесс (2) дает заметный вклад в скорость убыли O2( a 1 Δ ) также в разрядной зоне ЭГСК в случаях когда имеется избыток атомов О. В [12] за счет удаления избытка атомов О получена рекордная плотность О2( а 1∆) в ЭГСК.
ЗАКЛЮЧЕНИЕ
Таким образом, процесс (2) предсказывает наблюдаемые скорости деактивации O2( a 1 Δ ) как в послеразрядной [3], так и в послефотолизной [5] зонах. Процессы (2)-(5) также необходимо включать в кинетическую схему процессов разрядной зоны ЭГСК. В ряде работ [4-9] приведены экспериментальные факты в пользу процесса (2), тогда как процесс (1) феноменологический и он не объясняет темпы убыли O2( a 1 Δ ) в послефото-лизной зоне [4, 5]. Скорость деактивации O2( a 1 Δ ) может быть уменьшена удалением избытка атомов O, например, добавлением в смесь NO [1] или покрытием стенок камеры окисью ртути [12], а также добавлением в смесь на выходе ЭГСК тушителей O3( υ ), таких как СО2, SF6, SiF4 и т.д.
Работы в СГАУ поддержаны Минобрнауки РФ в рамках программы повышения конкурентоспособности СГАУ на 2013-2020 гг. и Государственного задания вузам и научным организациям в сфере научной деятельности, (гос. задание № 3.161.2014/K), а в СФ ФИАН поддержаны РФФИ (грант № 14-05-97013).
Список литературы Кинетическая модель деактивации О2 ( а 1D)
- Benavides G.F., Woodard B.S., Zimmerman J.W., Palla A.D., Day M.T., King D.M., Carroll D.L., Verdeyen J.T., Solomon W.C. IEEE J. Quantum Electr. 48, 741 (2012).
- Mezhenin А.V., Azyazov V.N., IEEE J. Quantum Electr., 49, 739 (2013).
- Vasiljeva A.N., Klopovskiy K.S., Kovalev A.S., Lopaev D.V., Mankelevich Y.A., Popov N.A., Rakhimov A.T., Rakhimova T.V. J. Phys. D: Appl. Phys. 37, 2455 (2004).
- Azyazov V.N., Mikheyev P.A., Postell D., Heaven M.C. Chem. Phys. Lett. 482, 56 (2009).
- Azyazov V.N., Heaven M.C. Int. Jour. Chem. Kinet. (2014) в печати.
- Azyazov V.N., Mikheyev P.A., Heaven M.C. Proc. SPIE 7751, 77510E (2010).
- Steinfield J.I., Adler-Golden S.M., Gallagher J.W. J. Phys. Chem. Ref. Data 16, 911 (1987).
- Kurylo M.J., Braun W., Kaldor A., Freund S.M., Wayne R.P. J. Photochem. 3, 71 (1974).
- Rawlins W.T., Caledonia G.E., Armstrong R.A. J. Chem. Phys. 87, 5209 (1987).
- Клоповский К.С., Ковалев А.С., Лопаев Д.В., Рахимов А.Т., Рахимова Т.В. Физика плазмы 18, 1606 (1992).
- Zimmerman, J.W., Dissertation, University of Illinois at Urbana-Champaign
- Braginsky O.V., Kovalev A.S., Lopaev D.V., Proshina O.V., Rakhimova T.V., Rakhimov A.T., Vasilieva A.N. J. Phys. D: Appl. Phys. 40, 6571 (2008).
