Клиническая протеомика: новые диагностические возможности масс-спектрометрии

Автор: Миргородская О.А., Манойлов А.В., Торопыгин И.Ю., Козьмин Ю.П., Краснов Н.В., Самус Н.Л., Новиков А.В., Бубляев Р.А.
Журнал: Научное приборостроение @nauchnoe-priborostroenie
Рубрика: Масс-спектрометрия для биотехнологии. Интерпретация данных, методология, применение
Статья в выпуске: 4 т.18, 2008 года.
Бесплатный доступ
Разработан масс-спектрометрический метод идентификации сывороточного амилоида А (САА) - характерного маркера для диагностика рака яичника. Его определение осуществлено непосредственно в сыворотке крови человека без предварительного фракционирования с использованием регулируемого трипсинолиза. Разработаны подходы к количественному определению содержания этого белка в сыворотке крови по содержанию триптического фрагмента (68-87) САА, присутствующего в гидролизатах. Разработан масс-спектрометрический метод определения α2-макроглобулина (α2-М) в сыворотке и плазме крови человека. С использованием разработанного метода проанализировано содержание α2-М в сыворотке крови при раке яичника и плазме крови при болезни Альцгеймера. Показано, что концентрация α2-М существенно возрастает при раке яичника и уменьшается при болезни Альцгеймера.
Короткий адрес: https://sciup.org/14264567
IDR: 14264567 | УДК: [577.1:
Clinical proteomics: new diagnostic possibilities of mass spectrometry
A mass spectrometric method of serum amyloyd A (SAA) identification was developed. SAA is a protein, characteristic marker for ovarian cancer diagnostics. Its determination was accomplished directly in the human blood serum by regulated trypsinolysis without a preliminary separation. New approaches to quantitative determination of this protein in blood sera with using of concentration of its tryptic fragment (67-87) SAA, present in hydrolysates, were developed. Besides mass spectrometric method of determination of α2-macroglobulin (α2-M) in blood sera and plasma were developed. Concentrations of α2-M in blood sera under ovarian cancer and in blood plasma under Alzheimer disease were measured using developed methods. It was shown that concentration of α2-M significantly increases under ovarian cancer and decreases under Alzheimer disease relatively of donor's blood sera.
Текст научной статьи Клиническая протеомика: новые диагностические возможности масс-спектрометрии
ВВЕДЕНИЕ В качестве стандартов используются аналогич-
В настоящее время масс-спектрометрия находит все более широкое использование в медицине. Благодаря масс-спектрометрии возникло новое направление исследований в медицине — клиническая протеомика, которая занимается инвентаризацией белков человека, кодируемых его генами, и таким образом дополняет геномные исследования. При всей индивидуальности геномных карт именно состав белков определяет функциональное назначение каждой клетки, а также отражает реализацию возможностей проявления имеющихся наследственных или приобретенных генетических дефектов.
В качестве особого раздела клинической протеомики следует выделить выявление изменений компонентного состава белков при возникновении онкологических, сердечно-сосудистых, неврологических и других заболеваний. Как правило, именно изменение концентрации отдельных белков указывает на появление патологии и может быть использовано в диагностических целях. В связи с этим интенсивно развиваются подходы, которые позволяют наряду с идентификацией осуществлять количественное определение белка с использованием масс-спектрометрии [1-5].
В основе большинства известных масс-спектрометрических методов количественного определения белков лежат два процесса:
-
• гидролиз белков с использованием специфических ферментов, главным образом трипсина;
-
• добавление стандарта и масс-спектрометрический анализ.
ные триптические фрагменты, но отличающиеся по молекулярной массе за счет содержания в них стабильных изотопов. Из интенсивностей изотопного распределения смеси анализируемого вещества и стандарта вычисляется количественное соотношение, на основании которого может быть оценено относительное содержание белка или его абсолютное значение, если концентрация стандарта известна. Таким образом, возможность использования масс-спектрометрии для количественных измерений обусловлена доступностью индивидуальных стандартов для каждого анализируемого соединения.
В настоящей работе использована предложенная ранее [6] универсальная технология получения стандартов, основанная на изотопном обмене 16О на 18О в карбоксильных группах пептидов и белков. Эта технология позволяет получать стандарты, не меняя химической структуры, не только из любых индивидуальных карбоксилсодержащих соединений, но и их смесей. Источником таких смесей могут служить, например, триптические гидролизаты белков. С использованием этого подхода проведено измерение относительного содержания сывороточного амилоида А (САА) — характерного маркера онкологических заболеваний для сыворотки крови больных раком яичников.
Также в работе проведено измерение относительного содержания в сыворотке или плазме крови трех функциональных белков: двух основных ингибиторов — а1-антитрипсин (а1-ИП) и а2-макроглобулин (а2-М), а также плазминогена (Пг) — одного из основных компонентов фибринолитической системы крови. Отметим, что со- держание этих белков изменяется при возникновении патологических состояний и может быть использовано в диагностических целях.
РЕЗУЛЬТАТЫ ЭКСПЕРИМЕНТА И ИХ ОБСУЖДЕНИЕ
Традиционно при выборе сывороток или плазмы крови в качестве источника для масс-спектрометрического анализа отдельных белков осуществляется предварительное фракционирование. Это обусловлено тем обстоятельством, что изменяемая часть белков составляет существенно меньшую часть на фоне высоких концентраций отдельных белков в биологической пробе. Так, например, в плазме или сыворотке крови — материалах, наиболее часто используемых для анализа, — около 90 % белкового состава приходится на альбумин, и поэтому без предварительного фракционирования и полном трипсинолизе сложно выявить появление или убыль каких-либо белков на таком фоне [7].
В настоящей работе в отличие от традиционной пробоподготовки предлагается использовать регулируемый трипсинолиз белков сыворотки крови для последующего масс-спектрометрического анализа.
Для эксперимента использовались три образца сыворотки: 417 — здоровой пациентки и 411, 431 — двух пациенток с раком яичника. К образцам сывороток добавляли раствор трипсина из расчета, чтобы его молярная концентрация была ниже концентрации α 1 -ИП и несколько выше, чем α 2 -М. Через 1.5 ч реакцию останавливали добавлением трифторуксусной кислоты (ТФУ). Пробы анализировали с использованием MALDI-MS. Результаты масс-спектрометрической детекции представлены на рис. 1.
Анализируя полученные данные по трипсинолизу в выбранных условиях, прежде всего следует отметить следующее:
-
• гидролизаты всех сывороток не содержат триптических фрагментов, альбумина;
-
• количество триптических фрагментов существенно больше в сыворотке больных по сравнению со здоровой, и их интенсивности существенно выше "фона";
-
• в раковой сыворотке присутствует ряд новых продуктов, представленных в масс-спектре интенсивными ионами.
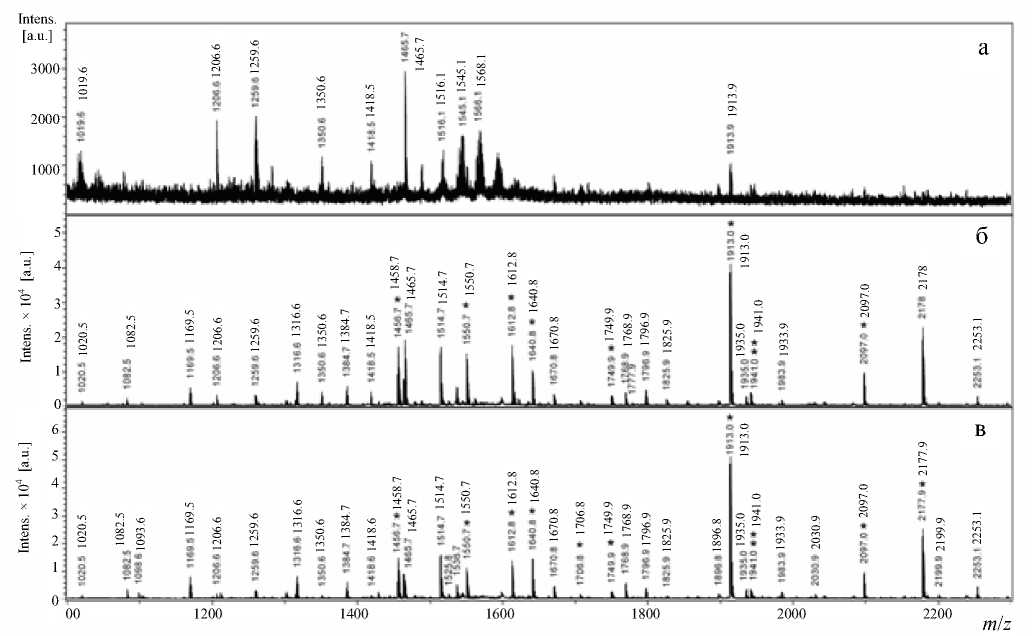
Рис. 1. Масс-спектры сыворотки в норме (а), раковой сыворотки 411 (б) и 431 (в) через 2 ч 15 мин трипсинолиза при 37 оС. (Аппаратные надписи на спектрограммах продублированы для разборчивости)
1706.8=
1670.8
RSFFSFLGEAFDGARDMWRAYSDMREANYIGSDKYFHARGNYDAAKRGPGGVWAAEAISD
=1550.7
100 104
1456.7
2097.0
ARENIGRFFGHGAEDSLADQAANEWGRSGKDPNHFRPAGLPEKY
2178.0
1640.8
1913.0
Рис. 2. Аминокислотная последовательность белков САА и регистрируемые в них масс-спектрометрически триптические пептиды
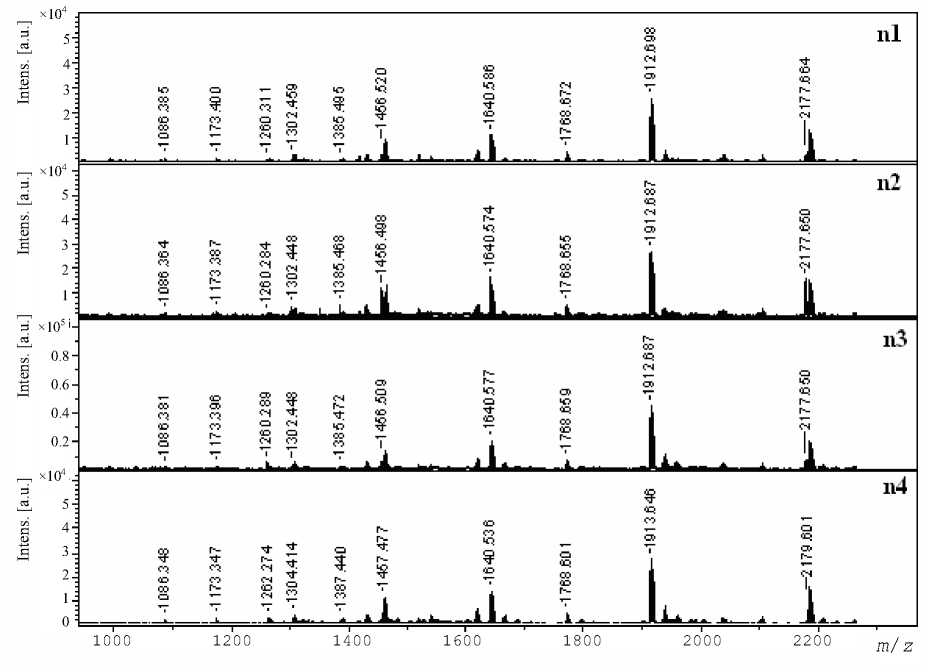
Рис. 3. Масс-спектры сывороток онкологических больных (n2– n4) после ограниченного протеолиза и в смеси с сывороткой n1, выбранной в качестве стандарта
По результатам поиска в МАСКОТ в образцах раковых сывороток 411 и 431 был с высокой достоверностью идентифицирован белок САА (score составлял 117 и 143, а перекрывание аминокислотной последовательности составляло 77 и 83 % соответственно). Пики, соответствующие триптическим пептидам САА, отмечены на рис. 1 звездочками. Аминокислотная последовательность белка САА и зарегистрированные пептиды представлены на рис. 2.
Таким образом, обнаружена способность трипсина в комплексе с α 2 -М селективно гидролизовать САА, что позволяет использовать ограниченный трипсинолиз в сочетании с масс-спектрометрией для выявления этого белка в сыворотке крови.
Фрагмент САА (91–104) m = 1640.81
Фрагмент САА (88–104) m = 1912.97
Фрагмент САА (68–87) m =2177.91
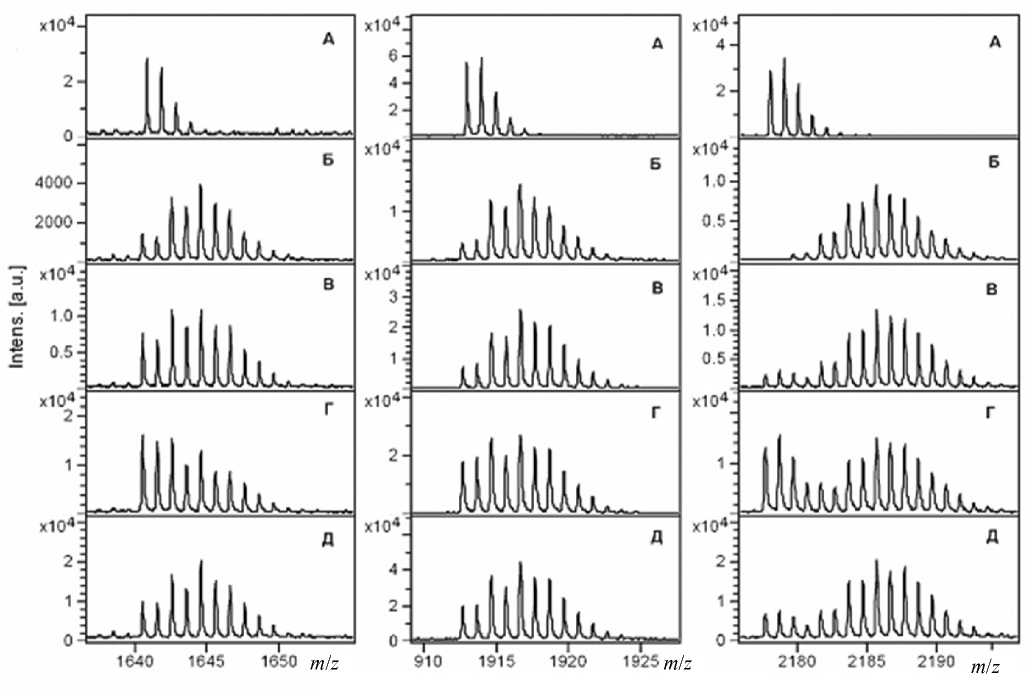
Рис. 4. Фрагменты масс-спектров сыворотки 411 после (А) и до (Б) изотопного обмена, смесей стандарта и раковых сывороток, смешанных в соотношении 5:1: 303 (В), 349 (Г) и 400 (Д)
Определение относительных концентраций САА в раковых сыворотках
Идентификация САА в крови пациентов всегда указывает на наличие патологии, причем тем более серьезной, чем выше концентрация этого белка. В связи с этим представляло интерес масс-спектрометрически не только обнаружить этот белок, но и определить его концентрацию. Для этого достаточно определить концентрацию одного триптического фрагмента САА, поскольку она соответствует содержанию исходного белка. Стандарты для определения САА получены из триптического гидролизата раковой сыворотки 411. Для этого гидролизат высушен и перерастворен в среде, подходящей для изотопного обмена. Полученный из гидролизата сыворотки 411 стандарт смешали с другими триптическими гидролизатами сывороток 303, 439 и 400 (соответствуют n2, n3, n4 на рис. 3)
и провели масс-спектрометрический анализ этих смесей. Масс-спектры таких смесей представлены на рис. 3.
Отметим, что во всех сыворотках присутствуют одинаковые триптические фрагменты. В данном случае сигналы каждого пептида достаточно широкие, поскольку в них смесь со стандартом, в результате чего имеет место более широкое распределение.
Для анализа сывороток использованы 3 пептида с m / z 1640.81, 1912.97 и 2177.91, аминокислотные последовательности которых представлены на рис. 2. На рис. 4 показаны фрагменты масс-спектров трех пептидов, выбранных для количественных расчетов в качестве стандартов сыворотки 411 (А), этих же пептидов до обмена и пептидов в смесях гидролизатов других сывороток 303, 349 и 400 (n2–4 на рис. 3).
Табл. 1. Относительная интенсивность пептидов в гидролизатах сывороток разных пациенток
|
n |
Serum |
Пептиды — фрагменты САА |
Относительная скорость гидролиза |
Соотношения концентраций САА/СААст in % |
||
|
(91–104), m / z = = 1640.81 |
(88–104), m / z = = 1912.97 |
(68–87), m / z = = 2177.91 |
(91–104) / (88–104) |
(68–87), m / z = 2177.91 |
||
|
1 |
411 (ст) |
— |
— |
— |
— |
100 |
|
2 |
303 |
13 |
2 |
6 |
6.5 |
30 |
|
3 |
349 |
47 |
32 |
35 |
1.7 |
175 |
|
4 |
400 |
17 |
11 |
15 |
1.5 |
75 |
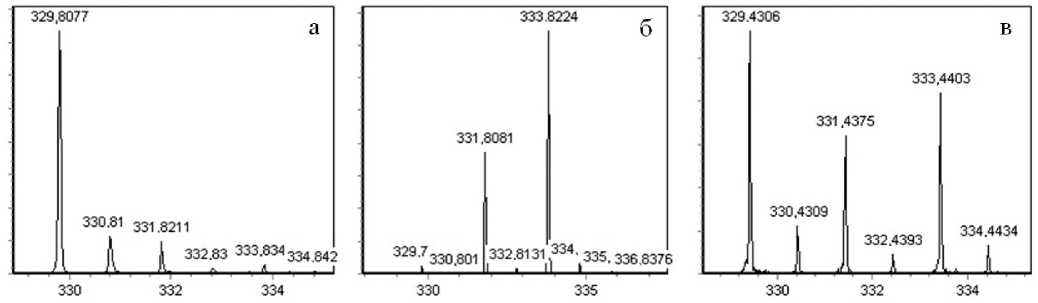
Рис. 5. Масс-спектры N-(тозил)-аргинина (ТА).
а — исходное вещество; б — стандарт ТА; в — смесь ТА с его стандартом
Рассчитанные относительные концентрации пептида (относительно стандарта) в сыворотках 303, 439 и 400 представлены в табл. 1.
Из этих пептидов только (68–87) CAA представляет собой продукт предельного гидролиза и может быть использован для количественных измерений САА. Два других — (88–104) CAA и (91– 104) CAA — не могут быть использованы для этих целей, поскольку первый из них представляет собой промежуточный продукт при получении второго. И их соотношение в гидролизатах зависит от времени трипсинолиза. Процесс гидролиза связи внутри пептида (88–104) CAA протекает существенно медленнее из-за его структурных особенностей. Вместе с тем из соотношения интенсивностей именно этих двух пептидов можно рассчитать относительные значения скоростей непосредственно трипсинолиза по убыли пептида (91–104) САА и по накоплению пептида (88–104) САА. Полученные значения представлены в табл. 1. Как следует из этих результатов, относительная ско- рость, рассчитанная из соотношения концентраций пептидов (88–104) САА и (91–104) САА, — наибольшая; но близкая наблюдается для сывороток 349 и 400 и наименьшая — для 303. Поскольку гидролиз осуществляется в одинаковых условиях включая концентрацию вносимого трипсина, относительные скорости отражают относительное содержание в сыворотках α2-М, поскольку чем выше концентрацая α2-М, тем выше активность трипсина [7]. Таким образом, из полученных данных можно судить об относительном содержании α2-М, изменения концентрации которого, по данным литературы [8–11], также отражает наличие и степень патологии.
Количественное масс-спектрометрическое определение α 2 -М по индуцированной триптической активности в сыворотке и плазме крови
Определение содержания α 2 -М в плазме и сыворотке крови осуществлялось по сохранению
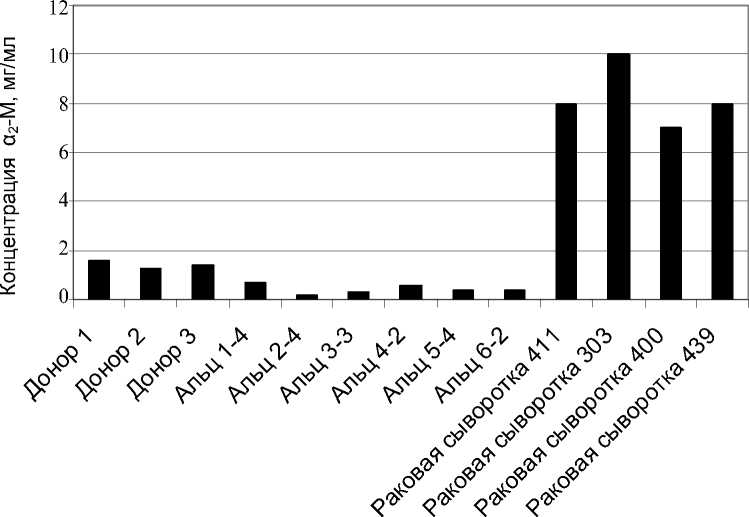
Рис. 6. Концентрации α 2 -М в сыворотках в норме, болезни Альцгеймера и при раке яичника (Альц 1-4, 2-4,…, 6-2 — идентификаторы проб)
активности трипсина после его введения в плазму в концентрациях, несколько меньших, чем возможное содержание α 1 -ИП. При этом трипсин прежде всего количественно связывается с α 2 -М с сохранением активности по отношению к метиловому эфиру N-тозил-аргинина (ТАМЕ). Остатки трипсина необратимо ингибируются α1-ИП. Определение трипсина в комплексе с α 2 -М осуществлялось с использованием ВЭЖХ и масс-спектрометрически по скорости превращения ТАМЕ в N-(тозил)-аргинин (ТА). Для масс-спектрометрического контроля за процессом гидролиза путем изотопного обмена был получен стандарт ТА. Масс-спектры исходного ТА, его стандарта, а также их смеси, представлены на рис. 5. В выбранных условиях обмена степень обмена в стандарте составляет 1.6. Из соотношения интенсивностей изотопного распределения смеси оценивалась степень превращения субстрата, которая составляет 29 % за 15 мин при 20 ºС.
Для определения α2-М в сыворотки вносился трипсин в тех же концентрациях что и при определении концентрации САА в сыворотках (предыдущий именованный подраздел). После образования комплекса сыворотки с трипсином полученные растворы смешивали с ТАМЕ и определяли скорости его гидролиза по отношению к скоро- стям гидролиза ТАМЕ трипсином в известной концентрации. Содержание α2-М определяли исходя из концентрации активного трипсина. При этом принимали во внимание тот факт, что содержание активного трипсина в исходном препарате порядка 40 %. Полученные таким образом концентрации α2-М приведены на рис. 6.
В результате проведенного эксперимента было обнаружена пониженная концентрация α 2 -М в плазме крови носителей болезни Альцгеймера (в 2–5 раз по сравнению с сывороткой крови доноров) и повышенная концентрация α 2 -М у больных раком яичника (в 4–6 раз).
МАТЕРИАЛЫ И МЕТОДЫ
Реактивы. Трипсин СПОФА (Чехия); метиловый эфир N-тозил-аргинина (TAME), M = 378.88 (Sigma, США); N-тозил-аргинин (ТА) фирмы Reannal (Венгрия), М = 328.8; гидрокарбонат аммония NH 4 HCO 3 (Sigma-Aldrich), M = 79.08; трифторуксусная кислота (ТФУ) (Merck, Германия), M = 114.0; муравьиная кислота (Merck, Германия), M = 46.03; ацетонитрил фирмы Вектон, M = 41.05.
Биологический материал. Были использованы сыворотки крови.
Табл. 2. Клинический материал
|
Носитель |
Заболевание |
|
Донор 1 (1980 г.р.)* |
Нет |
|
Донор 2 (1982 г.р.)* |
— ” — |
|
Донор 3 (1973 г.р.)* |
— ” — |
|
Альц 1-4 |
Болезнь Альцгеймера |
|
Альц 2-4** |
— ” — |
|
Альц 3-3** |
— ” — |
|
Альц 4-2** |
— ” — |
|
Альц 5-4** |
— ” — |
|
Альц 6-3** |
— ” — |
|
#303* |
Рак яичника |
|
#349* |
— ” — |
|
#400* |
— ” — |
|
#411* |
— ” — |
* Примечание. Биологич**еский материал:
* — сыворотка крови, ** — плазма крови.
Хроматографирование проводили на приборе Ми-лихром А-02 при температуре 35 ºС, градиенте (НСООН 0.25 % + H 2 O), (НСООН 0.25 % + СH 3 CN) 0–12 мин — 0–100 % при скорости потока 200 мкл/мин. Объем аликвоты при проведении хроматографии — 20 мкл.
Масс-спектрометрия. Масс-спектрометрические измерения проводили на времяпролетном масс-спектрометре МХ-5310, с ортогональным вводом ионов и оборудованном электрораспыли-тельным источником ионов (ESI-o-TOF) (Институт аналитического приборостроения РАН, Санкт-Петербург). Все спектры получены в режиме съемки положительных ионов. Объем пробы — 10–50 мкл, скорость подачи пробы — 1– 5 мкл/мин.
МALDI масс-спектры были получены на приборе Ultraflex (Bruker Daltonics, Bremen, Германия) в НИИ БМХ РАМН им В.Н. Ореховича (г. Москва). Все спектры получены в режиме съемки положительных ионов. В качестве матрицы использовалась α-циано-4-гидроксицинаминовая кислота.
Получение стандарта N-(тозил)-аргинина. Навеску 4.7 мг тозил-аргинина растворили в смеси 50 мкл H218O (соотношение 18О/16О — 50 %) и 3 мкл ТФУ. Обмен проводился в течение 3 ч при 50 ºС. После проведения обмена соотношение 16O/118O/218O стало равным 2/38/60, степень вклю- чения метки — 1.57. Для определения концентрации стандарта раствор смешивали с раствором исходного ТА точной концентрации и регистрировали масс-спектр смеси. После чего с помощью специального алгоритма [6] вычисляли соотношение концентраций исходного ТА и его стандарта.
Определение концентрации α 2 -M. Были приготовлены реакционные смеси: 20 мкл сыворотки (1/5), 10 мкл трипсина 0.08 мг/мл и 70 мкл буфера. После 15 мин комплексообразования по 4 мкл этих смесей были смешаны со смесью 10 мкл раствора ТАМЕ (2 мг/мл) и 36 мкл буфера. Реакция проходила в течение 15 мин. После чего реакцию останавливали разбавлением в 10 раз 0.2 %-й муравьиной кислотой. Для проведения масс-спектрометрических измерений полученную смесь смешивали с раствором стандарта ТА с соотношением ТАМЕ до реакции / С (стандарта ТА) 1/0.4. При этом учитывалось, что ТАМЕ с исходной концентрацией 2 мг/мл разбавлялся в 50 раз. Итоговая концентрация сыворотки в реакционной смеси — 1/312.5. Помимо этого стандартным образом была проверена активность трипсина (20 мкл раствора трипсина 0.001 мг/мл были смешаны с 10 мкл ТАМЕ 2 мг/мл и 20 мкл буфера, время реакции — 15 мин). Концентрацию α 2 -макроглобулина определяли следующим образом:
С (a2-M) = V (sera) X C (tryPsin) x M( a 2 - M )
v (trypsin) x C (sera) x M (trypsin) x 2
При этом учитывалось, что одна молекула α 2 -макроглобулина способна образовывать комплекс с двумя молекулами трипсина.
