Комбинированный безэталонный электрохимический метод определения ртути в водных растворах

Автор: Аверяскина Е.О., Ермаков С.С.
Журнал: Научное приборостроение @nauchnoe-priborostroenie
Рубрика: Исследования, приборы, модели и методы анализа
Статья в выпуске: 4 т.16, 2006 года.
Бесплатный доступ
Предложен комбинированный безэталонный (не требующий градуировки прибора по стандартным растворам и применения метода добавок) метод определения ртути в водных растворах, основанный на закономерностях инверсионной вольтамперометрии и потенциостатической кулонометрии.
Короткий адрес: https://sciup.org/14264464
IDR: 14264464 | УДК: 543.253
Combined calibration-free electrochemical method for determination of mercury in aqueous solutions
The paper offers a combined standardless (requiring no calibration against standard solutions and additives techniques) method for determination of mercury in aqueous solutions based on the principles of inversion voltammetry and potentiostatic coulometry.
Текст научной статьи Комбинированный безэталонный электрохимический метод определения ртути в водных растворах
Среди загрязняющих окружающую среду веществ, наиболее негативно влияющих на экосистемы и человека, ртуть занимает одно из первых мест. Она обладает высокой токсичностью, подвижностью, способностью накапливаться в трофических цепочках водных и континентальных биоценозов и признана одним из наиболее опасных глобальных загрязнителей окружающей среды.
В настоящее время для определения содержания ртути в различных средах (воздух, вода, почвы, продукты питания и т. д.) наибольшее распространение нашел метод атомно-абсорбционной спектроскопии (ААС) в "холодных" парах [1]. Основными недостатками метода являются недостаточно высокая воспроизводимость результатов анализа и необходимость применения относительно дорогостоящей аппаратуры [2, 3]. Дополнительные возможности для снижения пределов обнаружения при прямом определении следов ртути в воздухе открыл новый вариант дифференциальной ААС [4], но при этом еще больше возросла стоимость анализатора, что делает этот метод малодоступным для многих экоаналитических лабораторий. В то же время известно, что одним из наиболее чувствительных методов определения ртути в водных средах является инверсионная вольтамперометрия (ИВА) с использованием золотого электрода [5].
Своей высокой чувствительностью ИВА прежде всего обязана наличию в схеме анализа стадии предэлектролиза, т. е. электролитического концентрирования вещества на электроде. Стадию электролиза включает в себя и другой аналитический метод — кулонометрия при контролируемом потенциале (ККП). Последний не обладает чувствительностью ИВА, но не требует градуировки прибора, т. е. построения градуировочных кривых или применения метода добавок. Одним из условий корректного применения ИВА является постоян- ство концентрации определяемого вещества С0 в объеме раствора во время проведения измерений. Обычно это достигается использованием достаточно большого объема анализируемого раствора и малой площадью рабочего электрода. Последнее обстоятельство обеспечивает низкое значение тока предэлектролиза, которое соответственно не приводит к значительному изменению объемной концентрации определяемого вещества на стадии его электролитического концентрирования.
Другой электрохимический метод, включающий стадию электролиза при постоянном потенциале, — потенциостатическая кулонометрия наоборот основан на проведении измерений в условиях изменения концентрации аналита во времени, поскольку в ходе электролиза раствора происходит превращение определяемого вещества под действием электрического тока. Поэтому в кулонометрии используют электроды большой площади, а объем анализируемого раствора пытаются минимизировать.
Схематично условия проведения эксперимента в ИВА и ККП можно сформулировать следующим образом.
ИВА:
С ( t ) = C 0 = const;
id = id0 = const;(1)
-
i d0 = KC 0;
-
V/S → ∞.(2)
ККП:
С ( t ) = С 0 e – kt ;
id(t) = id0e– kt;(3)
i d ( t ) = K С ( t );
-
V/S → 0.(4)
Здесь С 0 — концентрация определяемого вещества в растворе; i d 0 — предельный диффузионный ток, отвечающий объемной концентрации С 0; i d ( t ) — предельный диффузионный ток в момент времени t ; t — время электролиза; V — объем раствора; S — площадь электрода.
Для повышения чувствительности и экспресс-ности определения аналитов методом ККП предложено использовать тонкослойные ячейки [6] (уменьшение объема раствора) и пористые электроды [7, 8, 9] (увеличение площади электрода). В этих работах электролиз раствора проводился до постулируемого авторами полного выделения вещества, и далее рассчитывалось его количество по закону Фарадея.
Полное выделение (превращение) вещества из раствора на электроде не является обязательным условием проведения кулонометрического анализа. Это было показано Мейтсом [10], который предложил находить Q ∞ , т. е. заряд, необходимый для полного выделения веществ, расчетным методом по формуле
Q = Q 2 - Q 1 Q 3
” 2 q 2 - ( Q i + Q 3 ) ,
где Q 1 , Q 2 , Q 3 — заряды, найденные интегрированием тока за отрезки времени t 1 , t 2 , t 3 соответственно, и
( t 2 - t i ) = ( t з - 1 2 ). (6)
Определение заряда и использование формулы Мейтса при концентрациях определяемого вещества менее 10–5 М затруднено, поскольку величины предельных диффузионных токов становятся меньше тока заряжения двойного электрического слоя, что не позволяет выделить фарадеевскую составляющую суммарного тока.
Предложено несколько вариантов инверсионных методов, позволяющих проводить безэталон-ное электрохимическое определение аналитов в растворе [11–16]. Авторы работ [11, 12] используют потенциометрический метод анализа, создавая с помощью предэлектролиза на поверхности электрода амальгаму определяемого металла известной концентрации, и далее по потенциалу системы Me(Hg)/Me n p- + p рассчитывают концентрацию аналита в растворе.
Ягнером [13] предложен инверсионно-хроно-потенциометрический вариант безэталонного электрохимического метода, основанный на многоциклическом растворении металла из амальгамы, полученной на стадии предэлектролиза и последующем нахождении заряда Q ∞ , отвечающего полному выделению вещества из раствора. Далее по закону Фарадея находится количество аналита в растворе.
Аналогичный метод, названный авторами "динамической кулонометрией", позволяющий учитывать величину тока заряжения двойного электрического слоя предложен в работах [14–15].
Комбинированный электрохимический метод определения свинца в водных растворах, основанный на закономерностях инверсионной вольтамперометрии разработан авторами работы [16].
Настоящая работа посвящена изучению возможности создания аналогичного предложенному в [16] комбинированного метода, сочетающего достоинства ИВА и ККП для определения ртути в водных растворах, проводя ИВА-измерения трижды с временами накопления, отвечающими условию (6), а затем находить Q ∞ по формуле (5). Решению этой проблемы на примере ИВА-опре-деления ионов ртути (II) в водных растворах посвящена настоящая работа.
ПРИНЦИП МЕТОДА
Идея комбинированного метода состоит в нахождении полного количества вещества, содержащегося в анализируемом растворе, с использованием ИВА-измерений и формулы Мейтса [10]. Это можно сделать следующим образом.
Сначала проводится электролиз на пленочно-золотом электроде при времени накопления Δ t 1, затем металл растворяется с поверхности золотого электрода, регистрируется ток растворения определяемого металла и находится заряд Q 1 интегрированием анодного тока по времени, затраченному на растворение ртути. Аналогичная процедура проводится при временах накопления Δ t 2 и Δ t 3 , и находятся соответствующие количества электричества Q 2 и Q 3 , причем обязательно должно выполнятся условие (6). Временнáя диаграмма измерений представлена на рис. 1. Для измерений использовали линейную развертку потенциала, для того чтобы иметь возможность проинтегрировать ток растворения по времени.
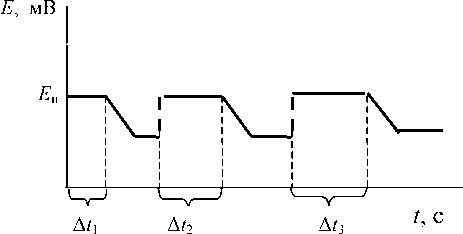
Рис. 1. Временнáя диаграмма изменения потенциала в комбинированном электрохимическом методе
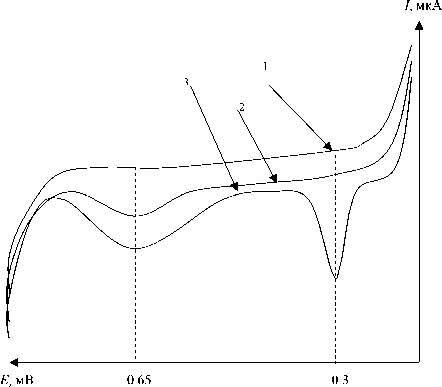
Рис. 2. Характерная вольтамперограмма ионизации ртути, полученная на золотом пленочном электроде в постояннотоковом режиме.
1 — кривая соответствует времени накопления 2.5 мин в фоновом электролите; 2 — кривая соответствует времени накопления в том же электролите со ртутью 2.5 мин; 3 — кривая соответствует времени накопления ртути 5 мин
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Инверсионно-вольтамперометрические измерения проводили на полярографе ПУ-1 (ЗИП, Гомель ) с датчиком АКВ-07 ( НПКФ "Аквилон", Москва ) и вольтамперометрическом анализаторе АКВ-07 МК с программным обеспечением Polar-3.0 (НПКФ "Аквилон", Москва ) в постояннотоковом и переменнотоковом режимах. Стандартный раствор ртути (II) с концентрацией 10–3 моль/л готовили по точной навеске Hg(NO 3 ) 2 ∙H 2 O. Стандартизацию полученного раствора проводили методом осадительного титрования [18]. Использовали реактивы квалификации х. ч., растворы готовили на бидистилляте.
Измерения проводили на золотом и пленочно-золотом электродах, скорость вращения электрода на стадии накопления составляла 1000 об/мин. В качестве фонового электролита использовали раствор состава 1М HCIO4 + 0.1M HCI [19]. Пред-электролиз в ходе ИВА-измерений осуществляли при потенциале –0.3 В, электрод сравнения — хлоридсеребряный. С целью получения электрохимической ячейки с регулируемым объемом в стандартную ячейку — стеклоуглеродный тигель объемом 50 мл — с помощью герметика по центру вклеивали цилиндр из полиэтилентерефталата диаметром 20 мм и высотой также 20 мм. Даль- нейшее регулирование объема осуществляли с помощью вкладышей из стеклоуглерода. Таким образом удается изменять рабочий объем ячейки от 5 до 1 см3. Измерения в больших объемах (20 мл) проводились в стандартной ячейке. Золотые пленочные электроды готовили электролитически нанесением пленки золота на стеклоуглеродные электроды с площадью 0.071 и 0.385 см2 , что соответствовало диаметрам рабочей поверхности электродов 3 и 7 мм соответственно.
РЕЗУЛЬТАТЫ ЭКСПЕРИМЕНТА И ИХ ОБСУЖДЕНИЕ
С целью выбора условий проведения эксперимента были сняты вольтамперограммы на золотом пленочном (см. рис. 2) электроде при различных концентрациях Hg2+ и временах накопления, что привело к следующим результатам.
При относительно небольших (~ 2.5 мин.) временах накопления на вольтамперограммах наблюдается 1 пик с потенциалом Е = + 0.65±0.01 В. По мере увеличения времени накопления (или концентрации) на вольтамперограммах появляется 2-й пик при Е = +0.30±0.01 В. Появление второго пика при менее положительных потенциалах, по всей вероятности, связано со следующими обстоятельствами. Поскольку коэффициенты диффузии ртути в золоте не высоки, то за время проведения эксперимента только небольшая часть осажденного жидкого металла успевает продиффундировать в толщу золота. Поэтому на поверхности золотого электрода появляется пленка ртути, не растворенной в золоте. При анодном окислении ртуть, не-растворенная в золоте, растворяется более легко, т. е. при менее положительных потенциалах. Ртуть, растворенная в золоте, ионизируется с большими энергетическими затратами, т. е. при более положительных потенциалах. Для доказательства этого предположения был проведен следующий эксперимент. Ртуть при разных временах накопления осаждалась на инертную подложку, в которой она не растворяется (в качестве такого материала был выбран стеклоуглерод). Затем проводилось растворение ртути с поверхности подложки. Экспериментально установлено, что ионизации ртути отвечает один пик при потенциале +0.3 В. Этот факт доказывает ранее сделанное предположение о том, что пик, наблюдаемый при +0.3 В на золотом электроде, отвечает ионизации ртути, нерастворенной в золоте. Кроме того, высоты обоих пиков, наблюдаемых на золотых электродах, зависели от концентрации ртути в растворе и от времени накопления, что также доказывает предположение о том, что оба пика на вольтампе-рограммах связаны с ионизацией ртути.
Высота пика, отн. ед.
. 1 ■ '
-
8J ■
-
0 J ■ --1--------------------------------1--------------------------------1-------------------------------- 1--------------------------------11-------------------------------- 1--------------------------------1-------------------------------- 1--------------------------------1-------------------------------- 1-----------------------------
0 2 4 6 8 1012
Время накопл. t н , мин
Рис. 3. Зависимость высоты пика ионизации ртути от времени накопления.
C Hg = 5 ∙ 10 –9 M; V = 3 мл; S эл = 0.071 см 2 ; поляро-граф ПУ-1; золотой пленочный электрод; переменнотоковый режим
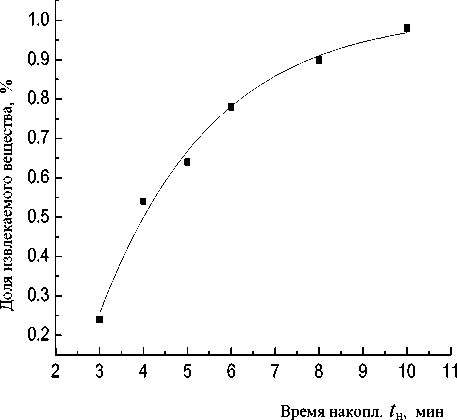
Рис. 5. Зависимость доли извлекаемого вещества от времени электролиза.
C Hg = 5 ∙ 10 –8 M; V = 6 мл; S эл = 0.071 см 2 ; переменнотоковый режим
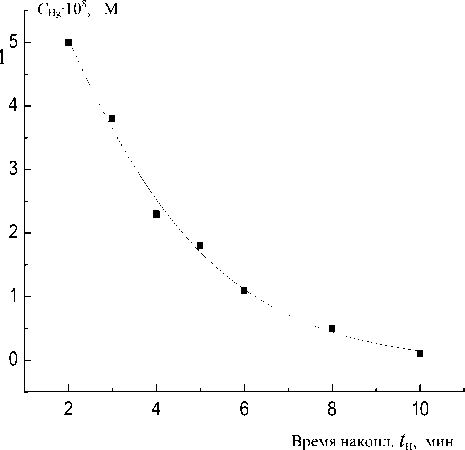
Рис. 4. Зависимость остаточной концентрации ртути (II) от времени накопления.
C оHg = 5 ∙ 10 –8 M; V = 6 мл; S эл = 0.071см 2 ; переменнотоковый режим
Для проверки выполнения условия (1), т. е. для подтверждения прямой пропорциональности величины аналитического сигнала объемной концентрации Hg2+ были проведены ИВА-измерения в растворах фонового электролита, содержащего ионы ртути в диапазоне концентраций 2∙10–8– 1∙10–6 М, в режимах ИВА с квадратно-волновой и линейной развертками потенциала. По результатам измерений была установлена прямо пропорциональная зависимость высот пиков на вольтам-перограммах (квадратно-волновой режим), площадей пиков и отвечающих им зарядов (линейная развертка) от концентрации Hg2+. Измерения проводились как при обычных для ИВА (V= 20 мл), так и при малых объемах растворов (V= 3 мл). Время накопления составляло 2 мин.
Изучение зависимости аналитического сигнала от времени накопления t н привело к следующим результатам (см. рис. 3). До t н = 10 мин зависимость практически линейна, а после t н = 12 мин высота пика перестает зависеть от времени. Этот результат хорошо согласуется с тем, что при электролизе раствора с высокой эффективностью перемешивания примерно за 12 мин происходит практически полное выделение вещества на электроде [10].
Для установления скорости изменения концентрации во времени был проведен следующий эксперимент. В растворе фонового электролита, содержащего 5∙10–8 М Hg2+, проводился электролиз при потенциале накопления, время накопления составляло 2 мин. Растворение осуществляли в раствор, не содержащий ионы Hg2+. По высоте пика ионизации ртути устанавливали его концентрацию в растворе. Зависимости изменения объемной концентрации ртути (II) и доли извлекаемого вещества от времени электролиза представлены на рис. 4 и 5 соответственно.
Доля извлекаемого вещества рассчитывалась по формуле
П =

Важнейшей величиной, характеризующей условия проведения измерений, в кулонометрии [17] является кулонометрическая константа k (см. условие (3))
DS k=V- ’
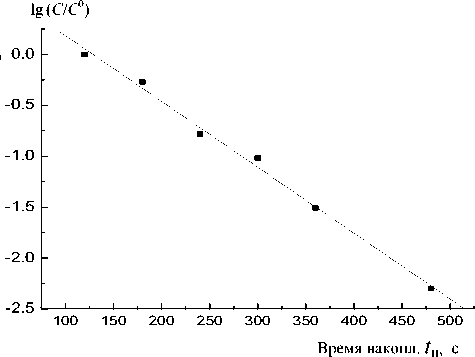
Рис. 6. Зависимость lg ( C / C 0 ) от времени накопления, полученная из данных, представленных на рис. 4
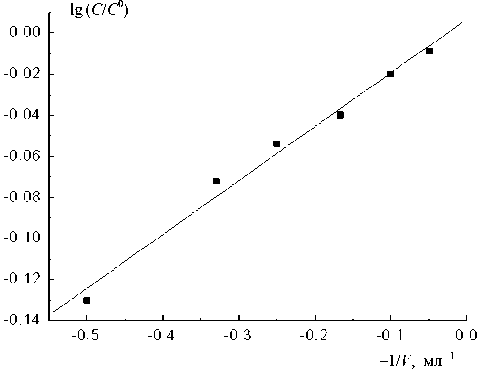
Рис. 7. Зависимость изменения концентрации от –1/ V.
Золотой пленочный электрод; Sэл = 7.1 мм2; CHg = =5∙10–8 M; постояннотоковый режим; tн = 5 мин где D — коэффициент диффузии, δ — толщина диффузионного слоя. Тогда скорость изменения концентрации во времени (см. условие (3)) будет выражаться уравнением
C = C 0 . e - kt = C 0 . e ( - DS/V S ) . (8)
Из уравнения (8) следует, что константа k может быть найдена из зависимости lg C / C 0 от t . На рис. 6 представлены данные рис. 4 в координатах lg C / C 0— t . Из них следует, что эта зависимость в данном случае представляет собой прямую линию, отвечающую уравнению lg C / C 0 = –kt , что выполняет условие (3). Величина константы, рассчитанная из данных, приведенных на рис. 6, составила: k Hg = 6.5 - 10-3 c-1.
Как следует из уравнения (8), при проведении электролиза раствора величина концентрации при определенном значении t будет зависеть от объема. Получена зависимость доли извлекаемого вещества С / С 0 при одном и том же времени накопления от объема раствора, который варьировался от 1 до 20 мл. При работе с объемами раствора больше 6 мл практически не происходит изменения объемной концентрации раствора. Очевидно, что наиболее приемлемо работать с ячейками объемом 1, 2, 3 мл. Построение этой зависимости в координатах lg C / C 0—1/ V приводит к получению прямолинейной зависимости (рис. 7), что хорошо согласуется с уравнениями (7) и (8).
С целью проверки предлагаемого метода были проведены следующие эксперименты. В постояннотоковом режиме были сняты инверсионные вольтамперограммы в растворах, содержащих фоновый электролит с известной концентрацией ионов ртути (II). Измерения проводились с временами накопления, отличающимися на равные промежутки времени. Нахождение Q 1 , Q 2 и Q 3 , соответствующих данным промежуткам времени, осуществлялось методом весового интегрирования. Далее по формуле (5) рассчитывали количество электричества, необходимое для электропревращения всего вещества, находящегося в растворе. Затем по закону Фарадея, зная объем раствора, находили концентрацию ионов ртути в растворе.
Были проанализированы растворы с концентрациями ионов ртути от 5∙10–8 М до 1∙10–6 М. В таблице приведены значения Q м и рассчитанных из них концентраций, полученных при временах накопления 5, 7.5, 10 мин для раствора с концентрацией 5 ∙ 10–8 М. Объем анализируемого раствора в ячейке составлял 1, 2 или 3 мл; измерения проводились на электроде с диаметром рабочей поверхности 3 мм.
Для растворов с более высокими концентрациями ионов ртути — 5 ∙ 10–7 М и 1 ∙ 10–6 М —
Результаты анализа растворов, содержащих ионы ртути, при разных временах накопления (время накопления в минутах приведено нижним индексом величины С )
|
№ измерения |
Введенная концентрация, С ∙108 М |
Объем раствора V , мл |
Q ∞ (теор), мкКл |
Найденное Q ∞ , мкКл |
Найденная концентрация, С ∙108 М |
|
1 |
2.0 |
4.0 |
15.4 |
14.7 |
С 2-4-6 = 1.9 |
|
2 |
″ |
″ |
″ |
15.1 |
С 4-6-8 = 2.0 |
|
3 |
″ |
″ |
″ |
16.7 |
С 6-8-10 = 2.2 |
|
4 |
″ |
″ |
″ |
16.6 |
С 8-10-12 = 2.2 |
|
5 |
5.0 |
4.0 |
38.6 |
37.4 |
4.9 |
|
6 |
″ |
″ |
″ |
38.7 |
5.0 |
|
7 |
″ |
″ |
″ |
38.0 |
4.9 |
инверсионно-вольтамперометрические измерения проводились с использованием рабочего электрода с диаметром 7 мм и при меньших временах накопления — от 0.5 до 2.5 мин.
Для проверки из значения Q ∞ , рассчитанного по формуле (8), найдено значение объемной концентрации раствора, которая получилась равной С 0 = = 4.98 ∙ 10–8 М, что очень хорошо согласуется с аттестованной величиной 5 ∙ 10–8 М для исследуемого раствора. Полученные результаты подтверждают перспективность комбинирования ИВА и КПП.
