Концепция таксономического пространства и интегральная оценка сдвигов в структуре микробных сообществ по данным анализа библиотек гена 16S-рРНК

Автор: Першина Е.В., Дольник А.С., Пинаев А.Г., Лошакова К.А., Андронов Е.Е.
Журнал: Сельскохозяйственная биология @agrobiology
Рубрика: Молекулярная идентификация структуры микробиомов
Статья в выпуске: 4 т.48, 2013 года.
Бесплатный доступ
Проблема таксономической структуры и динамики сложных микробиомов животных, человека, растений, почвы — одна из наиболее интригующих в современной микробиологии. Технологии высокопроизводительного секвенирования при изучении разнообразия микробных сообществ по полиморфизму генов 16S-рРНК позволили существенно увеличить объемы получаемых метагеномных данных, однако их корректный анализ и биологическая интерпретация вызывают затруднения, в частности в связи с эффектом избирательной амплификации с универсальными праймерами и собственно атрибутированием образцов. Для решения описанных проблем мы создали специальную операционную среду — таксономическое пространство (ТП), в котором последовательности гена 16S-рРНК представлены точками, геометрические расстояния между которыми соответствуют генетическим дистанциям между соответствующими последовательностями. Картирование данных по биоразнообразию гена 16S-рРНК в предлагаемом ТП и представление микробного сообщества как надорганизменной системы с присущими ей интегральными параметрами имеет ряд преимуществ перед традиционными подходами к оценке биоразнообразия. Так, в рамках ТП, где каждая последовательность гена 16S-рРНК получает собственный идентификатор из 42 координат, можно анализировать неатрибутированные последовательности в любой ампликонной библиотеке. Хотя описанное в работе ТП в строгом смысле не является многомерным математическим пространством (в частности, оси ТП взаимозависимы), полученные крайне высокие коэффициенты корреляции попарных генетических дистанций между последовательностями и их геометрическими аналогами, безусловно, свидетельствуют в пользу обоснованности применения ТП на практике. Развитие концепции ТП имеет большое значение не только при анализе структуры микробных сообществ, но и для изучения эволюционной истории гена 16S-рРНК. Поскольку модель позволяет дать описание любому его варианту (реализованному и еще не реализованному в ходе эволюции), в ее рамках могут решаться вопросы, связанные с происхождением и дивергентной эволюцией прокариотных таксонов (например, возможно определение гипотетического центра его происхождения, и тогда ТП реорганизуется в эволюционное пространство). В качестве экспериментальных объектов нами использованы разные почвенные микробиомы, в том числе смоделированы их изменения под воздействием условий среды (засоление). Однако предложенный в работе математический метод универсален и может быть использован для изучения не только биоразнообразия прокариот, но и сообществ эукариотических организмов (в том числе животных и растений; перспективным таксономическим маркером в этом случае будет ген 18S-рРНК).
Микробиом, почва, засоление, 16s ррнк, таксономическое про-странство
Короткий адрес: https://sciup.org/142133431
IDR: 142133431 | УДК: 579.6/.8:631.46:575.852''1
The concept of taxonomic space and integral estimates of shift in the structure of microbial community based on analysis of 16S rRNA gene libraries
The problem of taxonomic structure and dynamics of soil, plant animal and human microbiomes is one of the most intriguing in modern microbiology. High Performance Technologies sequencing of the 16S rRNA gene allows to get much more metagenomic data, but their correct analysis and biological interpretation are still complicated, in particular with regard to the effect of selective amplification with universal primers and proper attribution of the samples. To resolve the problems, we created a special operating environment, the taxonomic space (TS), in which the sequences of 16S rRNA gene are represented by dots, geometric distance between which corresponds to the genetic distance between the sequences. Mapping the 16S-rRNA gene biodiversity data in this TS and evaluation of the microbial community as overorganism, with its integral parameters, have a number of advantages if compared to the traditional approaches. Thus, in the TS where each sequence of the 16S rRNA gene gets its own identifier of the 42 coordinates, the unattributed amplicons in any PCR-library can be analyzed. Although the described TS is not strictly a multi-dimensional mathematical space, in particular, its axes are interdependent, an extremely high correlation coefficients, obtained for genetic distances between sequences and their geometric counterparts, unconditionally, testify in favor of the validity of the use of TS in practice. The development of TS concept is of great importance not only in the analysis of the structure of microbial communities, but also in imvestigation of 16S rRNA genes evolution. Since the model allows to give a description for any variant, both realized and not yet realized in evolution, the issues related to the origin and divergent evolution of prokaryotes may be investigated, for example, the hypothetical center of origin can be determine, and then the TS will become an evolutionary space. As a model, we used different soil microbiomes in which the changes were induced by environmental conditions (salinity), both natural and simulated. However, the application of this approach can be extended to other complex microbiomes, particularly the microbiota in animals. Moreover, the proposed mathematical method is universal and can be used to study not only biodiversity in prokaryotes, but also the communities of eukaryotic organisms, including animals and plants, with the 18S rRNA gene as a taxonomic marker.
Текст научной статьи Концепция таксономического пространства и интегральная оценка сдвигов в структуре микробных сообществ по данным анализа библиотек гена 16S-рРНК
При исследовании разнообразия микробных сообществ наиболее популярен анализ полиморфизма генов 16S-pPHK (1). Технологии высокопроизводительного секвенирования позволили значительно увеличить объемы экспериментальных данных по биоразнообразию, в результате чего стало возможным изучение не только структуры, но и динамики микробных сообществ (2, 3). Однако это поставило ряд проблем, связанных с получением, анализом и биологической интерпретацией метагеномных данных (4). Рассмотрим две из них.
Первая проблема обусловлена эффектом избирательной амплификации последовательностей 16S-pPHK при проведении мультиматричной ПЦР с универсальными праймерами (5, 6). Особенно ярко он проявляется при исследовании местообитаний с большой долей минорных групп микроорганизмов (таких как почвенное сообщество) и может привести не только к ПЦР-зависимым искажениям данных о структуре микробиома, но и к полному отсутствию ряда нуклеотидных последовательностей в ПЦР-про-дукте. Так, G.T. Bergmann с соавт. (7) показали, что из-за сравнительно слабого сродства всех современных конструкций универсальных праймеров с последовательностями бактерий из филы Verrucomicrobia данные по обилию этих микроорганизмов в почве были довольно сильно занижены (7). Несмотря на то, что проблема избирательной праймер-зависимой амплификации молекул 168-рРНК в той или иной степени затрагивает любое современное исследование, до сих пор не было предложено эффективных способов ее решения.
Вторая проблема появляется при анализе таксономической структуры микробных сообществ и ее биологической интерпретации. Основная трудность в случае метагеномных данных связана с высокой степенью биоразнообразия микробных сообществ, а также с наличием в их составе таксономически не идентифицируемых микроорганизмов. Доля микроорганизмов, не имеющих таксономической характеристики, на уровне как рода, так и более высоких таксонов (вплоть до филы) составляет, по разным оценкам, более половины от общего состава микроорганизмов в сообществе (8). Наличие таких форм не позволяет сравнивать состав микробиомов в независимых экспериментах и, в частности, ограничивает использование молекулярных методов в исследованиях, связанных с анализом большого числа образцов (микробиологическое картирование почв, мониторинговые исследования, биогеография микробиомов и др.).
Для решения описанных проблем мы создали специальную операционную среду для работы с нуклеотидными последовательностями — таксономическое пространство (ТП) для гена 168-рРНК. ТП представляет собой метрическое многомерной пространство, в котором последовательности гена 168-рРНК представлены точками, геометрические расстояния между которыми соответствуют генетическим дистанциям между соответствующими последовательностями (процент различающихся нуклеотидных позиций). Первая версия такого пространства описана ранее (9), но использованные тогда методы его построения не позволили получать высокие коэффициенты корреляции между генетическими дистанциями и их геометрическими аналогами ( r ® 0,3).
В настоящем исследовании предлагается альтернативный подход к построению таксономического пространства (ТП). В первой части нами будет продемонстрировано использование ТП для оценки эффекта избирательной праймер-зависимой амплификации в ПЦР на примере трех пар универсальных праймеров для гена 168-рРНК, во второй ТП будет использовано для оценки структуры и динамики микробных сообществ (в условиях засоления).
Методика. В качестве модели при оценке эффекта избирательной праймер-зависимой амплификации использовали образец дерново-подзолистой почвы, отобранный в июле 2007 года вблизи пос. Белогорка (Ленинградская обл.) из горизонта А1. При исследовании процесса природного почвенного засоления летом 2009 года провели экспедицию в район солончака Шингирлау (Казахстан) и вдоль градиента засоленности отобрали шесть образцов почвы (Т1, Т2, Т2-3, Т3, Т4 и Т5; содержание солей в Т1 — 1,23 %, в Т5 — 0,01 %). В 200 км от солончака были взяты три образца типичной для региона темно-каштановой почвы (одна целинная — НЦ и две залежные — НЗ и НК) (10). В опыте по засолению в почву с участка НК вносили раствор солей такого же состава, как в образце Т1 (на 100 г почвы Cl- — 8 mM, SO42- — 12 мМ, Na+ — 7 мМ, K+ — 0,5 мМ, Mg2+ — 2,5 мМ, Ca2+ — 10 мМ) до конечной концентрации 3 % (w/w). Соли каль ция и сульфатов добавляли раздельно. Эксперимент выполняли в двух тех нических повторностях (для контроля и опыта). В сосудах с почвой поддерживали постоянную влажность (60 % от полной влагоемкости). Пробы отбирали из трех равноудаленных друг от друга и от стенок сосуда точек и объединяли (в равных объемах) на стадии ДНК-экстрактов.
ДНК выделяли из почвенного образца (0,2 г) согласно описанию (11).
При оценке эффекта избирательной праймер-зависимой амплификации для получения библиотек гена 16S-рРHК были выбраны три пары универсальных праймеров: fD1/rD1 (27f: 5'-AGAGTTTGATCCTGGCTC-AG-3', 1525r: 5'-AAGGAGGTGATCCAGCC-3') (12); fBD1/rBD1 (642f: 5'-HAATHYGTGCCAGCAGC-3', 1445r: 5'-GTCRTCCYDCCTCCTC-3') (13) и Eu3 (63f*: 5'-AGGCCTAACACATGCAAGTC-3', 1494r: 5'-TACGGYTA-CCTTGTTACGAC-3') (14). Амплификацию выполняли по стандартной методике (11), полученные фрагменты клонировали в векторе pAL-TA («Евроген», Россия) по протоколу производителя и использовали конструк ции для трансформации компетентных клеток Escherichia coli (DH10B) (15). Для каждой пары праймеров была создана библиотека фрагментов гена 16S-рРHК (L1 — Eu3, L2 — fD1/rD1 и L3 — fBD1/rBD1). Последовательность нуклеотидов определяли с использованием праймера FGPS (485-292) 5'-CAGCAGCCGCGGTAA-3' (16) в автоматическом секвенаторе SEQ8000 с применением реагентов фирмы-производителя («Beckman Culter», США) в соответствии с прилагаемым протоколом. Выравнивание последовательностей проводили в программе Clustal X, построение мат риц генетических дистанций и филогенетического древа — в программе
MEGA 5. Таксономическую идентификацию последовательностей выпол няли на сервере RDPII (17).
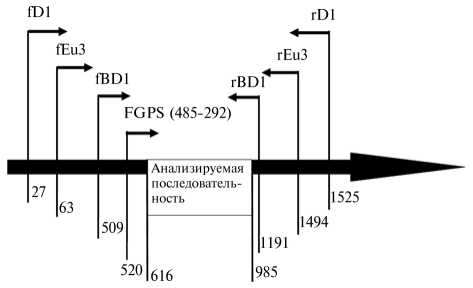
Рис. 1. Расположение трех пар универсальных праймеров относительно нуклеотидной последовательности гена 16S-рРНК Escherichia coli (штамм K12).
Для получения библиотек гена 16S-рРHК при оценке структуры и динамики микробных сообществ в условиях засоления амплификацию проводили с универсальными праймерами F515 и R806 к вариабельному участку V4 (18). Пиросеквенирование ампли-фикатов выполняли на приборе GS Junior («Roche», Швейцария) согласно рекомендациям производителя. Анализ последователь ностей осуществляли в программе QIIME v. 1.5.0 (19). Из нуклеотидных последовательностей удалили служебные последовательности, провели фильтрацию, выравнивание, построение матрицы генетических дистанций и таксономическую идентификацию с использованием параметров, установленных по умолчанию. Кластерный анализ образов выполняли с использованием алгоритма «unweighted unifrac».
Достоверность различий оценивали по t-критерию Стьюдента и F- критерию Фишера.
Результаты . Анализ таксономического состава ампликонных библиотек. Библиотеки гена 168-рРНК, полученные с использованием различных конструкций универсальных праймеров. Локализация последовательностей трех пар универсальных праймеров в гене 16S-pPHK, использованных для получения библиотек, приведена на рисунке 1.
Для анализа в каждой ампликонной библиотеке выбрали примерно равное число последовательностей: 33 — из L1, 29 — из L2 и 33 — из L3. Нуклеотидные последовательности были отнесены к бактериальным филам Proteobacteria (32 %), Acidobacteria (26 %), Verrucomicrobia (7 %), Actinobacteria (4 %), Bacteroidetes (3 %), Planctomycetes (3 %), Chlamydiae (3 %) и Firmicutes (2 %) и депонированы в GenBank (the National Center for Biotechnology Information — NCBI) с присвоением идентификаторов HQ412669-HQ412763.
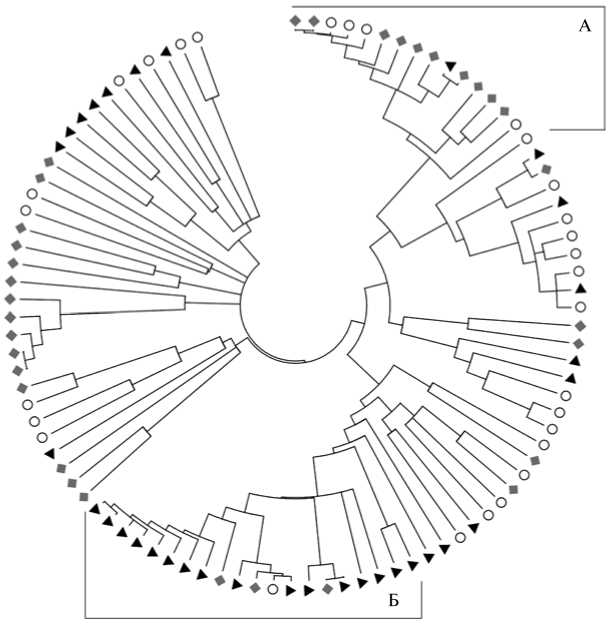
Рис. 2. Распределение последовательностей из ампликонных библиотек L1 (♦) , L2 ( о ) и L3 (▲ ) , полученных для гена 168-рРНК с использованием разных пар универсальных праймеров, на филогенетическом древе: А — последовательности из филы Proteobacteria , Б — последовательности из филы Actinobacteria .
При построении филогенетического древа было показано, что последовательности из трех библиотек неравномерно распределяются в пределах основных прокариотных таксонов (рис. 2). Так, в филе Proteobacteria основную часть составляли нуклеотидные последовательности из библиотеки L1. Более того, подавляющее большинство из них формировали компактную группу, родственную порядку Rhizobiales . В филе Acidobacteria сложилось противоположное распределение с перевесом в сторону последовательностей из библиотек L2 и L3. Причем последовательности из L2 кластеризовались преимущественно с ацидобактериями из групп Gp3 и
Gp2, а последовательности из L3 — с группой Gp1. Неравномерность в распределении нуклеотидных последовательностей наблюдалась также для фил Verrucomicrobia и Chlamydiae (табл. 1). Полученные данные наглядно
1. Таксономическая идентификация последовательностей гена 16S-pPHK при анализе ампликонных библиотек L1, L2 и L3, полученных с разными парами универсальных праймеров
|
Фила |
1 L1 | |
L2 |
1 L3 |
|
Proteobacteria |
18 |
8 |
7 |
|
Acidobacteria |
4 |
13 |
11 |
|
Verrucomicrobia |
— |
1 |
6 |
|
Actinobacteria |
4 |
— |
— |
|
Bacteroidetes |
2 |
— |
1 |
|
Planctomycetes |
— |
2 |
1 |
|
Chlamydiae |
— |
— |
3 |
|
Firmicutes |
2 |
— |
— |
|
Примечание. Прочерк означает, что филы не выявлен. |
представитель |
||
демонстрируют эффект праймер-зависимой амплификации, который проявился на уровне фил.
Оценка структуры и динамики микробных сообществ в условиях засоления. Засоление относят к числу наиболее мощных экологических факторов (20). Подробно динамика микробного сообщества при естественном засолении описана нами ранее (10). Здесь же мы остановимся на кратком сравнительном анализе структуры сообществ в условиях есте ственного и искусственного засоления. Во всех пробах были выявлены представители 21 бактериальной филы, среди которых доминировали бактерии из фил Actinobacteria, Bacteroidetes, Firmicutes, Proteobacteria. Сравни тельный анализ процессов естественного и искусственного засоления выявил ряд общих особенностей в изменении структуры сообществ. В обоих случаях при засолении возрастала доля бактерий из фил Firmicutes и Bacteroidetes . Изменения касались одних и тех же порядков (Bacillales и Sphingobacteriales) и семейств (Balneolaceae и Bacillaceae) (рис. 3).
Solirubrobacteraceae (Actinobacteria)
SOG AS31 HA
(Chloroflexi)
Rubrobacteraceae (Actinobacteria)
Bacillaceae (Firmicutes)
Balneolaceae (Bacterioidetes)
Streptomycetaceae (Actinobacteria)
Nocardioidaceae (Actinobacteria)
Pseudomonadaceae
(Gammaproteobacteria)
Enterobacteriaceae
(Gammaproteobacteria)
Rhodospirillaceae (Alphaproteobacteria)
Gemmatimonadetes, HA
Alicyclobacillaceae (Firmicutes)
Balneolaceae
(Bacterioidetes)
Actinobacteria. HA
Bacteria, HA
Actinobacteria. HA
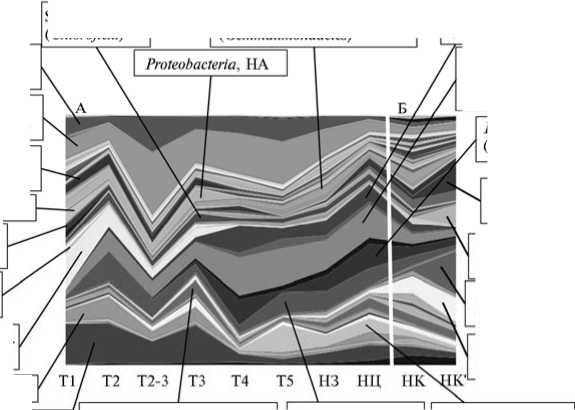
Рис. 3. Таксономическое разнообразие микробных сообществ (на уровне семейства) в условиях естественного (А, Т1-НЦ) и искусственного (Б, НК-НК') почвенного засоления, оцененное по данным секвенирования ампликонных библиотек гена 16S-pPHK: Т1-Т5 — образцы солончаковой почвы, НЗ, НЦ и НК — образцы темно-каштановой почвы (НЦ — целинная, НЗ и НК — залежные), НК' — образец НК после добавления солей того же состава, что в варианте Т1; НА — последовательности, не атрибутируемые на уровне порядка.
Также в сообществах происходила закономерная смена типичных почвенных актинобактерий (Rubrobacteriaceae и Solirubrobacteriaceae) на группы актинобактерий, более приспособленных к условиям засоления (не иден- тифицируемых на уровне семейства бактерий из класса Actinobacteria в случае естественного засоления и бактерий из сем. Nocardioidaceae и Strep-tomycetaceae в случае искусственного засоления (см. рис. 3).
Наряду с общими закономерностями в развитии процессов естественного и искусственного засоления нами были обнаружены и существенные различия. Внесение в почву солей вызвало значительное снижение разнообразия в сообществе. Состав фил Firmicutes и Bacteroidetes по сути ограничивался лишь двумя семействами — Bacillaceae и Balneolaceae . Засоление также привело к выраженному сокращению числа семейств актинобактерий (см. рис. 3, Б). Наиболее засоленный участок солончака, напротив, характеризовался большим разнообразием среди представителей названных фил (см. рис. 3, А). Наличие существенных различий в структуре микробных сообществ для сравниваемых процессов подтверждает также дендрограмма, где образцы Т1 и НК' находятся в разных кластерах (рис. 4).
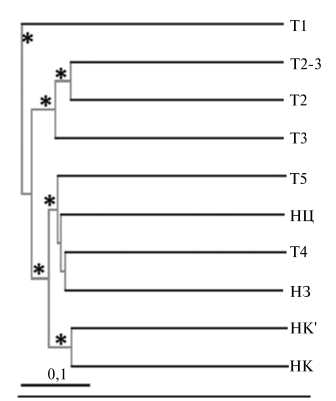
Рис. 4. Результаты кластерного анализа структуры микробных сообществ в условиях естественного и искусственного почвенного засоления (по данным секвенирования ампликонных библиотек гена 168-рРНК): Т1-Т5 — образцы солончаковой почвы, НЗ, НЦ и НК — образцы темно-каштановой почвы (НЦ — целинная, НЗ и НК — залежные), НК' — образец НК после добавления солей того же состава, что в варианте Т1 . Алгоритм кластеризации — «unweighted unifrac»; звездочкой отмечены кластеры со значением достоверности более 80 %.
Построение таксономического пространства и выбор оптимальной системы координат. Для оценки эффекта избирательной амплификации мы построили многомерное математическое пространство — таксономическое пространство для гена 168-рРНК
(ТП). Ранее (9) положение нуклеотидных последовательностей в ТП опре деляли в системе координат правильного симплекса. В настоящей работе в качестве координат ТП использовали систему реперных точек — после-
|
i |
j |
А |
В |
C |
|
|
i |
0 |
d# |
diA |
dig |
d;c |
|
j |
0 |
djA |
djB |
djc |
|
|
А |
0 |
dAB |
dAC |
||
|
в |
0 |
dsc |
|||
|
с |
0 |
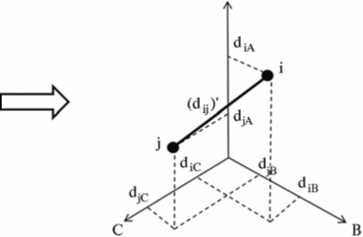
Рис. 5. Схема трансформации генетических дистанций в таксономические: i, j — нуклеотидные последовательности 168-рРНК, входящие в состав анализируемой ампликонной библиотеки; А, B, С — нуклеотидные последовательности, выбранные из базы данных Ribosomal Database Project (RDP) II для построения таксономического пространства (реперные точки), d — генетические дистанции между нуклеотидными последовательностями (p-distance — доля различающихся нуклеотидных позиций в последовательностях, %), d i ' — геометрические аналоги генетических дистанций).
довательностей гена 168-рРНК у отдельных представителей крупных бак териальных и археотных таксонов. Положение точки в ТП задавали прямой трансформацией генетических дистанций (доля различающихся нуклеотидных позиций, выраженная в p-distance) от реперных последовательностей в геометрические координаты. Такая система координат позволила рассчитать геометрические аналоги (dij') для генетических дистанций (dij) (рис. 5).
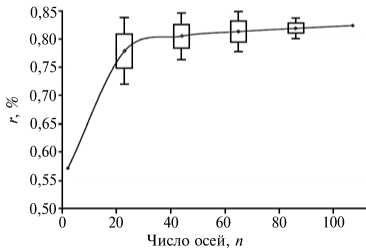
Рис. 6. Зависимость средних значений коэффициента корреляции от числа выбранных «координатных осей» — последовательностей гена 168-рРНК (всего 107) с разной представленностью среди основных бактериальных и археотных фил.
Для формирования оптимального набора координат ТП из наиболее крупной на сегодняшний день базы данных по гену 16S-рРHК — Ribosomal Database Project II отобрали 107 по следовательностей по критерию представленности среди основных бактериальных и археотных фил. Затем вычислили коэффициенты корреляции (геометрические расстояния) между матрицами генетических дистанций (dij) и их рассчитанных в ТП аналогов (dij') для всех возможных наборов координат (Сn 107, где n = 2, ..., 107). Очень высокое соответствие dij и dj наблюдалось при сравнительно небольшом числе координат — от 20 до 45 (рис. 6). Для дальнейших построений использовали 42 последовательности с наилучшими значениями r. Выбранные последовательности были равномерно распределены между 23 бактериальными и 2 археотными филами, при этом их значительная доля принадлежала к наиболее крупным бактериальным филам: Proteobacteria (7 последовательностей), Firmicutes (4 последовательности), Actinobacteria (3 последовательности) и Acidobacteria (3 последовательности). В список также вошли представители фил Bacteroidetes, Chlamydiae, Deinococcus-Thermus, Nitrospirae, Spirochaetes, Thermotogae, Chlorobi, Chloroflexi, Cyanobacteria, Gemmatimonadetes, Lentisphaerae, Planctomycetes, Verrucomicrobia, OP10, TM7, WS3, SR1, Euryarchaeota, Korarchaeota.
Исследование ампликонных библиотек в ТП. Оценка эффекта избирательной праймер-зависимой амплификации . В построенном таким образом ТП ампликонные библиотеки L1, L2 и L3 будут представлены в виде «облаков точек», а эффект избирательной амплификации выразится в различиях этих облаков по форме и положению в зависимости от варианта универсальных праймеров. При численной оценке эффекта избирательной амплификации выбрали наиболее простые параметры, описывающие взаимное расположение и геометрию ампликонных библиотек в ТП, — центральную точку (имеет средние координаты по каждой из координатных осей) и дисперсию (мера разброса точек относительно центра). Для характеристики различий в положении ампликонных библиотек в ТП определили расстояние между всеми парами центральных точек. Все обнаруженные нами различия в геометрии ампликонных библиотек были статистически достоверны согласно критериям Стьюдента и Фишера.
Для пары L1-L2 d = 0,09, t = 30, f = 33, для пары L1-L3 d = 0,10, t = 34, f = 36, для пары L2-L3 d = 0,07, t = 24, f = 29, где d — расстояние между центральными точками ампликонных библиотек, t — число осей с достоверными различиями в координатах центральной точки (по t-критерию Стьюдента), f — число осей с достоверными различиями в дисперсии (по F-критерию Фишера). То есть расстояния между центральными точками библиотек свидетельствовали в пользу сходства для L2 и L3 и их
2. Максимальные смещения координат центральной точки по осям в парах ампликонных библиотек гена 16S-pPHK для разных родов бактерий
|
Род |
| (L1-L2)|(L1-L3)|(L2-L3) |
||
|
Eubacterium |
0,011 |
0,019 |
0,007 |
|
Acidimicrobium |
0,000 |
0,016 |
0,016 |
|
Parachlamydia |
0,007 |
0,018 |
0,011 |
|
Simkania |
0,006 |
0,015 |
0,009 |
|
Holophaga |
0,039 |
0,041 |
0,002 |
|
Acidobacteria |
0,033 |
0,004 |
0,029 |
|
Xiphinematobacter |
0,014 |
0,033 |
0,020 |
|
Korarchaeota NA |
0,011 |
0,007 |
0,018 |
|
Thermotoga |
0,015 |
0,009 |
0,007 |
|
Roseomonas |
0,006 |
0,021 |
0,015 |
|
Caulobacter |
0,040 |
0,044 |
0,004 |
|
Sinorhizobium |
0,045 |
0,048 |
0,003 |
|
Azotobacter |
0,004 |
0,019 |
0,015 |
|
Campylobacter |
0,008 |
0,020 |
0,011 |
|
Примечание. |
Использованные |
при по- |
|
|
лучении библиотек L1, L2 и L3 пары универ- |
|||
|
сальных праймеров |
см. в разделе «Методика». |
||
принципиального отличия от L1. Иными словами, универсальные праймеры D1 и BD1 по сравнению с Eu3 в целом сходным образом описывали структуру анализируемого микробного сообщества. Такой вывод дополнительно подкрепляется историей их создания. Известно, что праймеры fD1/rD1 начали использовать для анализа микробного разнообразия одними из первых (12). Их аналогами (с учетом значительных изменений, произошедших в базах данных за более чем десятилетний период) стали праймеры fBD1/rBD1 (13). Праймеры Eu3 повсеместно применяются при T-RFLP (terminal restriction fragment length polymorphism) анализе микробных сообществ (14), но их особенность состоит в суще ственном перевесе в использованных при конструировании последова тельностей в сторону протеобактерий (21). Возможно, именно поэтому при анализе секвенированных последовательностей из библиотеки Eu3 было обнаружено большое число генов 168-рРНК ризобий — типичных представителей этой филы.
3. Расстояния (d) между центральными точками ампликонных библиотек гена 16S-pPHK, полученными для разных почвенных образцов в условиях естественного засоления
С целью более детального анализа различий в структуре библиотек была построена таблица смещений координат центральной точки по осям (смещение представляет собой разность двух координат центральных точек). Наибольшие смещения наблюдались для библиотеки L1 в филе Proteobacteria (координаты Si-norhizobium и Caulobacter ), наименьшие — в паре L2-L3 (здесь максимум отмечали для двух родов ацидобактерий — Xiphi-nematobacter и Acidobacteria NA) (табл. 2). Все обнаруженные точки смещения хорошо соответствуют данным, полученным с использованием традиционного подхода (построение филогенетического древа), где также было показано, что основные различия в структуре ампликонных библиотек связаны с филами Proteobacteria и Acidobacteria (см. рис. 2, табл. 1).
Оценка структуры и динамики микробных сообществ в условиях засоления с использованием интегральных параметров ТП. Как и в случае ампликонных библиотек L1, L2 и L3, ампликонные библиотеки, характеризующие процесс естественного и искусственного засоления в ТП будут представлены в виде «облаков точек». В этом случае их взаимное расположение в ТП (расстояние между центральными точками) служит мерой сходства или различия в структуре сообществ. При расчете таких расстояний между всеми парами в условиях естественного засоления было показано, что максимальных значений названный параметр достигает при сравнении образца из наиболее засоленного участка солончака (Т1) с незасоленной почвой (НЦ) (табл. 3).
Произведенные вычисления дисперсии точек относительно центральной точки и сравнение параметров среднего и дисперсии с использованием критериев Стьюдента и Фишера показали, что наблюдаемые в ТП различия достоверны. Поэтому мы можем использовать ТП при изучении динамики микробных сообществ.
Для ее описания были введены параметры вектора смещения (показывающего направление сукцессии) и угла между векторами смещения (меры сходства процессов сукцессии, происходящей в различных микробных сообществах). Направление вектора смещения для сообщества определяется разницей координат его начальной и конечной точек (центральных точек сообществ), модуль вектора равен расстоянию между центральными точками, угол между векторами рассчитывается по формуле скалярного произведения векторов.
При анализе процессов почвенного засоления в ТП построили три вектора: НЦ^Т1, НЗ^Т1 (развитие естественного засоления) и НК^НК' (развитие искусственного засоления) (рис. 7).
А
Б
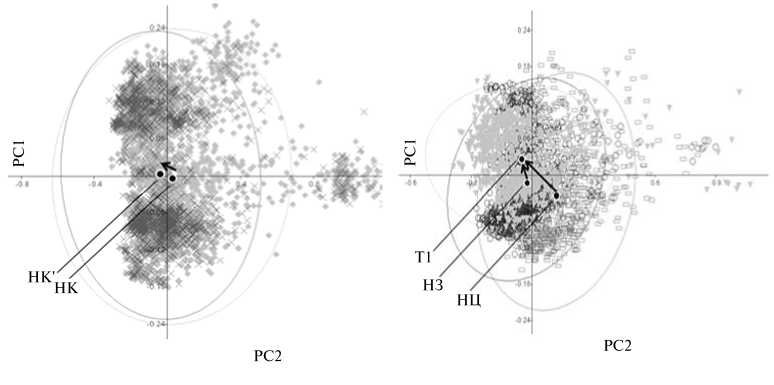
Рис. 7. Определение интегральных характеристик микробных сообществ в таксономическом пространстве (центральные точки и углы между векторами смещения для микробных сообществ) , построенном на основании ампликонных библиотек гена 16S-pPHK, для условий естественного (А) и искусственного (Б) засоления: Т1, НЗ, НЦ, НК, НК' — соответственно образцы почвы солончаковой, темно-каштановой (НЦ — целинная, НЗ и НК — залежные), НК' — образец НК после добавления солей того же состава, что в варианте Т1. PC1, PC2 — координаты проекции с максимальными различиями в расположении сравниваемых совокупностей точек.
При анализе углов между векторами в условиях естественного и искусственного почвенного засоления оказалось, что искомые значения лежат в пределах сектора 90° (42° для пары НК^НК'/НЦ^Т1 и 73° — для НК^НК'/НЗ^Т1). С учетом предположения, что угол в этой системе варьирует от 0° (полностью сходное развитие сообществ) до 180° (диаметрально противоположные изменения в структуре сообществ), полученные нами данные свидетельствуют в пользу наличия сходных паттернов в развитии исследуемых сообществ. Такой результат хорошо согласуется с полученным нами ранее при сравнительном анализе таксономической структуры метагенома, где, наряду с выявленными существенными различиями, 84
в засоленных образцах отмечалось появление бактерий, принадлежащих к одним и тем же порядкам и семействам.
Важно подчеркнуть, что тенденции, наблюдаемые при сукцессии в микробных сообществах, становятся очевидными только при подробном анализе их таксономической структуры и не могут быть выведены на основании результатов традиционно используемого для решения подобных задач кластерного анализа. Использование векторов смещения имеет и ряд других преимуществ перед традиционными методами, среди которых упрощение процедуры анализа динамики сообществ, возможность ее сопоставления у различных по природе сообществ и ранжирования экологических факторов по силе их воздействия на сообщество.
Понятно, что по результатам одного исследования невозможно однозначно трактовать значения углов между векторами развития микробных сообществ. Для более точной оценки нужно выполнить ряд исследований, в которых необходимо проанализировать факторы по воздействию на микробное сообщество: выделить факторы, оказывающие противоположное и однонаправленное действие, разделить факторы по силе воздействия на микробное сообщество, провести сопоставление величины угла с той или иной силой воздействия экологического фактора и т.д.
Очевидно, что успех представленной в настоящей работе версии ТП связан с перебором всевозможных комбинаций из множества «координатных осей». Так, было показано, что высоких коэффициентов корреляции можно достичь лишь при использовании нескольких десятков координат, в то время как предыдущая версия ТП (9) оперировала лишь 13 точками отсчета. Таким образом, предложенная альтернативная модель позволила выявить основную проблему, возникающую при конструировании ТП — проблему размерности. По всей видимости, в дальнейших работах необходим поиск определенного компромисса между описанными моделями.
Итак, мы показали возможность использования концепции таксономического пространства (ТП) при решении проблем, связанных с избирательной праймер-зависимой амплификацией гена 168-рРНК и изучением таксономической структуры микробных сообществ. Картирование данных по биоразнообразию гена 168-рРНК в ТП и представление микробного сообщества как надорганизменной системы с присущими ей интегральными параметрами не только дополняет традиционные подходы к оценке биоразнообразия, но и имеет перед ними ряд преимуществ. Например, в рамках концепции ТП разрешается наиболее актуальный на сегодняшний день вопрос о неатрибутированных последовательностях, поскольку в ТП каждая последовательность гена 168-рРНК получает собственный идентификатор, представленный набором из 42 координат, что позволяет обнаружить ее в любой ампликонной библиотеке. Мы полностью отдаем себе отчет в том, что описанное в работе ТП не является в строгом смысле многомерным математическим пространством (в частности, оси ТП взаимозависимы). Тем не менее, в исследовании были получены крайне высокие коэффициенты корреляции попарных генетических дистанций между последовательностями с их геометрическими аналогами, что, безусловно, свидетельствует в пользу концепции ТП. Очевидно, что успех представленной здесь версии ТП связан с перебором всевозможных комбинаций из множества «координатных осей». Продемонстрировано, что высоких коэффициентов корреляции удается достичь лишь при использовании нескольких десятков координат, что указывает на проблему размерности как основную при конструировании ТП. Дальнейшее развитие концепции ТП имеет большое значение не только для анализа структуры микробных сообществ, но и для изучения эволюционной истории гена 16S-pPHK. Поскольку модель позволяет дать описание любому варианту его структуры (включая как эволюционно реализованные, так и еще не реализованные), в ее рамках могут обсуждаться вопросы, связанные с происхождением и дивергентной эволюцией не только прокариот, но также одноклеточных и многоклеточных эукариот (при использовании гена lSS-рРНК для построения ТП) (например, возможно определение гипотетического центра происхождения таксона). В последнем случае ТП может быть реорганизовано в эволюционное пространство.
