Ковалентное допирование нитрида углерода карбазолом и бензохалькодиазолами: моделирование электронных свойств в альтернативных приближениях

Автор: Григорьева Е.А., Матвейчук Ю.В.
Журнал: Вестник Южно-Уральского государственного университета. Серия: Химия @vestnik-susu-chemistry
Рубрика: Физическая химия
Статья в выпуске: 1 т.16, 2024 года.
Бесплатный доступ
Представлены результаты теоретического анализа электронных свойств соединений на основе фрагментов нитрида углерода, состоящих из трех гептазиновых колец (мелона), ковалентно связанных с гетероциклическими заместителями: электрон-акцепторными 2,1,3-бензохалькодиазолами и электрон-донорным карбазолом. Моделирование выполнено на двух альтернативных уровнях: молекулярная газофазная модель и модель одномерного полимера с периодическими граничными условиями. Эти уровни позволили сравнить разность энергий граничных орбиталей молекулярной модели и ширину запрещенной зоны цепочечного полимера одного и того же соединения. Обнаружено, что, с одной стороны, селенсодержащие гетероциклы значительно больше, чем другие ковалентно связанные допанты рассмотренной серии, уменьшают разность энергий граничных орбиталей, а с другой стороны, допирование мелона только карбазолом максимально уменьшает ширину запрещённой зоны. При эффективном допировании, максимально уменьшающем разность граничных орбиталей, высшая занятая орбиталь локализована на электрон-донорном фрагменте молекулы, а низшая свободная - на электрон-акцепторном фрагменте молекулы. Также показано, что допированные мелоны формируют комплексы с бензиловым спиртом, образованные за счёт появления между ними нековалентных связей. При этом меньшая прочность таких связей в комплексах с мелоном, замещённым электрон-акцепторными селенсодержащими фрагментами, предполагает более эффективное окисление спирта в таких системах. Результаты моделирования адекватно соответствуют имеющимся экспериментальным данным по ширине запрещённой зоны мелона, допированного электрон-акцепторными молекулами. Быстрые газофазные расчёты подходят для качественной оценки зависимостей разности энергий граничных орбиталей от вида и количества вводимого допанта, а более затратные расчёты полимерных структур позволяют провести корректную оценку самих значений ширины запрещённой зоны полимеров нитрида углерода при введении различных допантов.
Нитрид углерода, мелон, электрон-донорные и электрон-акцепторные гетероароматические допанты, разность граничных орбиталей, ширина запрещённой зоны
Короткий адрес: https://sciup.org/147243265
IDR: 147243265 | УДК: 544.169 | DOI: 10.14529/chem240110
Covalent doping of carbon nitride with carbazole and benzochalcodiazoles: simulation of electronic properties in alternative approximations
The results of theoretical consideration of the electronic properties are presented for compounds based on carbon nitride fragments consisting of three heptazine rings (melon) covalently bonded with heterocyclic substitutes (electron acceptors like 2,1,3-benzochalcodiazoles and electron donors like carbazole). The simulation has been performed at two alternative levels: a molecular gas-phase model and a model of one-dimensional polymer with periodic boundary conditions. These levels have allowed comparing the energy change of frontier orbitals and the band gap of a chain polymer for the same compounds. It has been found that, on the one hand, the selenium-containing heterocycles reduce the energy change of frontier orbitals significantly more than other covalently bonded dopants of the considered series; on the other hand, the doping of melon with mere carbazole minimizes the band gap. At efficient doping, which minimizes the energy change of frontier orbitals the highest occupied orbital is localized on the electron-donor fragment of the molecule, and the lowest unoccupied orbital is localized on the electron-acceptor fragment of the molecule. It has also been shown that the doped melons form complexes with benzyl alcohol due to appearance of non-covalent bonds between them. Wherein, the lower strength of such bonds in complexes with melon, substituted with the electron-acceptor selenium-containing fragments, suggests more efficient oxidation of an alcohol in such systems. The simulation results appropriately correspond to the available experimental data of the band gap of melon doped with electron acceptor molecules. The fast gas-phase calculations are suitable for qualitative estimation of dependences of the energy change of frontier orbitals upon their type and the amount of covalently bonded dopant, whereas the more expensive calculations of polymer structures allow a correct estimation of the band gap values of carbon nitride polymers with various dopants.
Текст научной статьи Ковалентное допирование нитрида углерода карбазолом и бензохалькодиазолами: моделирование электронных свойств в альтернативных приближениях
Нитрид углерода вызывает широкий интерес исследовательского сообщества благодаря простоте получения, привлекательной для многих практических целей электронной структуре, высокой физико-химической стабильности и доступности [1–3]. Графитоподобный нитрид углерода g-C 3 N 4 (ГНУ) в целом представляет собой плоскую двумерную структуру, которая в стабильной кристаллической форме не зафиксирована [4]. Разновидности ГНУ образованы триазиновыми или гептазиновыми звеньями. Наиболее энергетически выгодную структуру имеет мелем, представляющий собой гептазиновое звено. Мономеры мелема, связываясь между собой через атомы азота, образуют мелон [5], который является важным промежуточным продуктом при конденсации меламиновых колец до нитрида углерода [6].
Особый интерес представляет дальнейшая модификация сопряженной π-системы полигептазиновых цепей. Исследование органических полупроводниковых полимеров получило развитие в течение последних двух десятилетий, что привело к возникновению новой отрасли электронных устройств на основе органических светоизлучающих диодов, OLED, полевых транзисторов, OFET и органических фотогальванических элементов, OPV [7, 8]. Это развитие основано на модификации электронной структуры высокосопряженных органических полимеров. Основным подходом к этой модификации является введение донорных и акцепторных заместителей в полимерные цепи, которые нагнетают или отнимают электронную плотность в сопряженной системе, тем самым влияя на энергетические уровни и ширину запрещенной зоны полимера.
Хотя этот подход оказался эффективным при создании органической электроники, он мало применяется в синтезе фотокатализаторов для расщепления воды, окисления или разложения загрязняющих веществ из-за низкой стабильности и фотокоррозии [9, 10] Мы предлагаем применить донорно-акцепторный подход к ГНУ, который оправдан из-за его более высокой стабильности и надежности. Более того, убедительное количество публикаций однозначно указывает на положительное влияние молекулярного допирования и сополимеризации с g-C3N4 на фотокаталитическую активность. Исследована сополимеризация с производными бензола: п-фенилендиамин [11], тригидроксибензол [12], терефталевая кислота [13, 14], пиромеллитовый ангидрид [15–17], аминобензойная кислота [18], пиридины [19, 20], пиримидины [21–24], триазол [25], 3-аминотиофен-2-кабонитрил [9, 26, 27], фенилмочевина [28] и бензотиадиазол [29, 30].
Однако в большинстве этих публикаций изучается влияние одного допанта на активность ГНУ, что затрудняет полное понимание преобразования электронной структуры. Хотя в некоторых отчетах [31, 32] и показано влияние ряда допантов на фотокаталитическую активность по отношению к выделению водорода, сложно сделать вывод о природе возникающей эффективности катализа.
Полупроводниковые свойства ГНУ обусловлены делокализованной π-электронной структурой мелемовых или триазиновых субструктур, состоящих из атомов углерода и азота в sp2-гибридизации. С одной стороны, ароматическая π-система ГНУ в чистом виде содержит внутренние недостатки, с практической точки зрения, такие как быстрое рекомбинирование разделённых зарядов, неполное поглощение видимой части спектра, существенным образом сдерживающие её фотохимический потенциал [33, 34]. С другой стороны, на большом числе примеров было показано, что внедрение малых ароматических и гетероароматических молекул в полимерную структуру ГНУ способствует оптимизации запрещённой зоны полупроводниковых материалов, настройке уровней ВЗМО и НСМО, улучшению светочувствительности и фоторазделению зарядов [35].
Важным сдерживающим фактором для понимания особенностей взаимодействия ГНУ с допантами и механизмов его каталитической активности является недостаточность данных о структуре объектов исследований [36]. Первыми приближениями на пути к моделированию ГНУ являются его структурные звенья – меламин, мелем, димелем, мелон [37]. Такие звенья возможно успешно смоделировать в газофазном кластерном приближении с помощью различных методов и подходов, учитывая известный опыт [38–40]. Несмотря на невозможность учёта межзвенных взаимодействий, слоевых эффектов, на этом уровне моделирования удаётся корректно описать особенности, связанные со свойствами ковалентных связей, и в первом приближении учесть влияние атомных и молекулярных допантов типа пиридина на электронные характеристики [41]. Однако прогностические модели, позволяющие предсказать каталитическую активность ГНУ как его главное целевое свойство, требуют прямого учёта ближнего порядка и симметрии. Это становится возможным только при переходе к моделям с периодическими граничными условиями, позволяющим учесть двумерную или трёхмерную периодичность [42]. Рассмотрение таких моделей в литературе встречается намного реже, однако в числе наиболее выдающихся стоит отметить модели, в рамках ТФП, периодических расчётов с использованием локализованных атомных базисных наборов в CRYSTAL [43], а также сравнение различных подходов к вычислению ШЗЗ [44].
Все вышеперечисленные модели затрагивают вопросы построения двух- и трёхмерной структуры без точно известной геометрии взаимного расположения звеньев, а также позволяют получить ряд сопоставимых с расчётом электронных характеристик.
В представленном исследовании мы поставили следующие цели:
-
• моделирование молекул мелона, допированных рядом гетероциклов с выраженными электрон-донорными и электрон-акцепторными свойствами в 2 приближениях: кластерном и с периодическими граничными условиями;
-
• расчет электронных характеристик полученных систем, анализ взаимосвязи рассчитанных характеристик и имеющихся экспериментальных данных;
-
• определение влияния допантов на энергию связывания замещённого мелона с молекулой бензилового спирта;
-
• анализ результатов расчетов в 2 приближениях и разработка рекомендации по возможности использования методик расчетов для модификации свойств допированного мелона.
В работе представлены результаты моделирования структуры и свойств рассматриваемых π-электронных систем с экспериментальными данными на двух уровнях моделирования, а именно: 1) оптимизированные структуры и энергия граничных орбиталей для изолированных молекул мелона, состоящих из трех и девяти гептазиновых звеньев, полученные путем кластерного расчета молекулы в газофазном состоянии; 2) рассчитанная ширина запрещенной зоны для полимерных цепей мелона, смоделированных из девяти гептазиновых звеньев как мономеров, с периодическими граничными условиями. В обоих случаях одна молекула легирующей примеси соответствовала одной молекуле мелона или полимерному звену.
Экспериментальная часть
Серия анализируемых молекулярных структур мелона ( Tm_M ), ковалентно-связанного с замещенными гетероциклами на основе карбазола и 2,1,3-бензохалькодиазола ( 1–20 ), формировалась путем замещения атомов водорода и чередования молекулярных допантов, представленных молекулами с выраженными как электрон-донорными, так и электрон-акцепторными свойствами (табл. 1). Кластерные квантово-химические расчеты, включающие локализацию равновесной геометрии допированных структур мелона, были выполнены с использованием ТФП, в программе Gaussian 16 [45]. Были использованы функционалы с базисными наборами B3LYP/6-311+G(d,p) и HSE06/DZVP. Учет эффектов сольватации произведен методом поляризационного континуума То-маси, PCM [46]. Критерий сходимости по энергии 10–8 ат. ед. и параметры по умолчанию уровня точности Tight. Выполнен расчет энергий граничных молекулярных орбиталей: высшей занятой (ВЗМО) и низшей свободной (НСМО), и разности граничных орбиталей, ΔE MO = Е НСМО – Е ВЗМО .
Таблица 1
Состав молекулярно-допированных структур мелона и энергии граничных орбиталей в кластерных расчётах, уровень расчета HSE06/DZVP
|
№ |
Обозначение |
Электрон-акцепторный допант |
Электрон-донорный допант |
E ВЗМО , эВ |
Е НСМО , эВ |
ΔE MO , эВ |
|
0 |
Tm_M |
– |
– |
–6,32 |
–2,75 |
3,57 |
|
1 |
Tm_O |
1,2,5-оксадиазол |
– |
–6,56 |
–3,09 |
3,46 |
|
2 |
Tm_S |
1,2,5-тиадиазол |
– |
–6,49 |
–3,01 |
3,48 |
|
3 |
Tm_Se |
1,2,5-селенадиазол |
– |
–6,47 |
–3,00 |
3,48 |
|
4 |
Tm_B_O |
2,1,3-бензоксадиазол |
– |
–6,40 |
–3,16 |
3,23 |
|
5 |
Tm_B_S |
2,1,3-бензотиадиазол |
– |
–6,23 |
–3,10 |
3,13 |
|
6 |
Tm_B_Se |
2,1,3-бензоселенадиазол |
– |
–6,13 |
–3,20 |
2,93 |
|
7 |
Tm_D |
– |
карбазол |
–5,64 |
–2,66 |
2,98 |
|
8 |
Tm_D_S |
– |
дибензотиофен |
–5,90 |
–2,74 |
3,16 |
|
9 |
Tm_O_D |
1,2,5-оксадиазол |
карбазол |
–5,75 |
–2,95 |
2,80 |
|
10 |
Tm_S_D |
1,2,5-тиадиазол |
карбазол |
–5,72 |
–2,88 |
2,84 |
|
11 |
Tm_Se_D |
1,2,5-селенадиазол |
карбазол |
–5,71 |
–2,87 |
2,85 |
|
12 |
Tm_B_O_D |
2,1,3-бензоксадиазол |
карбазол |
–5,72 |
–3,08 |
2,64 |
|
13 |
Tm_B_S_D |
2,1,3-бензотиадиазол |
карбазол |
–5,69 |
–3,03 |
2,65 |
|
14 |
Tm_B_Se_D |
2,1,3-бензоселенадиазол |
карбазол |
–5,68 |
–3,14 |
2,53 |
|
15 |
Tm_O_D_S |
1,2,5-оксадиазол |
дибензотиофен |
–6,00 |
–3,02 |
2,99 |
|
16 |
Tm_S_D_S |
1,2,5-тиадиазол |
дибензотиофен |
–5,97 |
–2,94 |
3,03 |
|
17 |
Tm_Se_D_S |
1,2,5-селенадиазол |
дибензотиофен |
–5,96 |
–2,93 |
3,04 |
|
18 |
Tm_B_O_D_S |
2,1,3-бензоксадиазол |
дибензотиофен |
–5,97 |
–3,12 |
2,85 |
|
19 |
Tm_B_S_D_S |
2,1,3-бензотиадиазол |
дибензотиофен |
–5,94 |
–3,07 |
2,87 |
|
20 |
Tm_B_Se_D_S |
2,1,3-бензоселенадиазол |
дибензотиофен |
–5,93 |
–3,18 |
2,75 |
Локализация равновесной геометрии одномерных полимерных структур 8 исследуемых образцов производилась с помощью программного пакета CRYSTAL17 [47] методом ТФП с периодическими граничными условиями следующим образом. В качестве мономерного звена взята молекула, состоящая из трёх параллельных трёхзвенных молекул мелона, соединённых между собой в шахматном порядке. То есть одно звено моделируемого полимера состояло из 9 соединённых между собой в три ряда молекул мелема. Затем один или два атома водорода на краях мономерного звена замещались на молекулярные фрагменты допантов в нескольких сочетаниях. Информация о мономерных звеньях, использованных для моделирования соответствующих полимеров, приведена в табл. 2. Было установлено, что применение к моделированию того же уровня расчёта, что и для молекулярных структур, а именно B3LYP/6-311+G(d,p), даёт завышенные и далёкие от экспериментальных [48] значения ШЗЗ. Поэтому для локализации равновесной геометрии полимеров был использован уровень расчёта HSE06/pob-DZVP_rev2 [49, 50], давший более близкие к эксперименту значения ШЗЗ для незамещённого полимера. Для релаксации как исходной структуры мелона, так и замещённых полимеров использована опция POLYMER программного пакета CRYSTAL17 и следующие значения параметров программы: TOLDEG 0.00001, TOLDEX 0.00003, TOLDEE 10, TOLINTEG 14 14 14 14 24. Полученные значения ШЗЗ приведены в табл. 2.
Таблица 2
Состав молекулярно-допированных структур мелона в расчётах полимеров, значения ШЗЗ и разности энергий граничных орбиталей для соответствующих молекул
|
№ |
Обозначение |
Допанты |
Структура |
ШЗЗ, эВ |
ΔEMO, эВ |
|
1 |
Tm_M |
– |
2,91 (2,56)*, (2,70)** |
3,57 |
|
|
2 |
Tm_B_Se |
2,1,3-бензоселенадиазол |
2,72 (1,88)* |
2,93 |
|
|
3 |
Tm_D |
карбазол |
2,26 |
2,98 |
|
|
4 |
Tm_B_Se_D |
2,1,3-бензоселенадиазол, карбазол |
2,49 |
2,53 |
|
|
5 |
Tm_B_O |
2,1,3-бензоксадиазол |
2,92 (2,41)* |
3,23 |
|
|
6 |
Tm_B_S |
2,1,3-бензотиадиазол |
2,69 (2,00)* |
3,13 |
|
|
7 |
Tm_B_S_D |
2,1,3-бензотиадиазол, карбазол |
2,45 |
2,65 |
|
|
8 |
Tm_B_O_D |
2,1,3-бензоксадиазол, карбазол |
2,60 |
2,64 |
Экспериментальное значение ШЗЗ [36]; ** Экспериментальное значение ШЗЗ [54].
Ан а л и з хара к тера в за и мод ействия в комплексах из молекулы ГНУ и б е н зи лов ого спирта п рои зв од и лся при п омощи ра счё т ов э н е рги и в з аимоде й с тв ия (E int ) молекул бензилового спирта и терм и н а л и з ов а н н ого фр агме н та н и три да у гле р од а. Р а сче т E int произведен методом противовеса, с у че том оши б к и с у п е рп ози ци и ба зи с н ого наб о ра п о фо рм у л е : E int = E AB – (E A + E B ) – E BSSE , где E AB – эн ергия м ол е к у лярн ого комплекса бензилового спирта и фрагмента н и три д а у глерод а, E A – эн ергия мол е к у лы бе н зил ов ого с п ирта , E B – энергия фрагмента нитрида углерода, E BSSE – п оп ра в к а н а ош и бк у с у перп оз и ц и и б а зи с н о го н а бора [ 51]. Расчет энергии нековалентных взаимодействий E HB п рои зве де н по уравнению Эспиноза с соавторами [ 52]: E HB = ΣE i,HB , E i,HB = 0,5υ(r b ), где E i,HB – эн ерги я отдельно взятого нековалентного взаимоде й с тв и я в к омп л е к се, υ(rb) – п л отн ос ть п оте н ц и альн ой э н е рги и в связевой критической точке (3,–1) этого взаимодейст в и я. Р а с чет вы п олн е н в програ мме Gaussian 16 [45], критерий сходимости по э нерги и 10–8 ат. ед. и п а раме тры п о у м ол ча ни ю у ро вня точности Tight. Визуализация и анализ ра сс чи та н н ых с т руктур молекул, пол и м еров, к омпл ексов и их молекулярных орбиталей выполнены п ри п омощ и п рограммы ChemCraft 1.8 [53].
Обсуждение результатов
Моле ку ля р ный дизай н к ов але нтн о -допированных соединений мелона
Для мод е ли р ов а н и я и золи рованных молекул мелона и их допированных п ро изводных были в ы б ра н ы ф ра гм е н ты, с остоящ и е из трех и девяти гептазиновых звеньев, из н а ча льн о и ме ющ ие п л ос к у ю с тру к ту ру. Рас п олож е н ие донорных и акцепторных гетероциклов в Г НУ в ыб ра н о с у ч е том и ме ющ и хс я э кс п е риме н та л ьн ых данных [36]. Разности энергий граничных молекулярных орбиталей ( ΔE MO ) д ля и золи рованных молекул мелона, замещенных од н и м и з бе н-зо[c ][1, 2, 5]ха л ьк о ге н ад и азолов , и оценки ШЗЗ для полимерных цепей тех ж е фрагме н тов , п олу ч е н ны е с п е ри о д и че ск и м и гра н и чными условиями, приведены в табл. 2. Также м ож но отметить, ч т о м ол ьн ое с оотн оше н и е молекулы допанта и мелона (в пересчете на мела ми н) в п оли ме рн о м зв е н е при ме рн о соответс т в уе т 5%-ному допированию в эксперименте. Этот факт позволяет срав н и ть э к с п е ри м е н та льные и ра с че тные э ле к трон ные харак те рис ти к и д оп и рованной молекулы ме- л он а , и х те нд е н ц и и и оц ени ть в озможные структурные особенности допирова н н ого мелон а .
С п е ц и фи че ск ое ра с поло же н ие допирующих заместителей повлияло на п рос транственн у ю структуру смоделированных сое д и не н и й ( ри с . 1 ) . Э лек трон-акцепторные допанты на основе 2,1,3- б е н зоха льк о д и а золо в могу т замещать атом Н первичной аминогруппы, т. е. располагаться в о д н ой из в е ршин гептаз и н ов ого к ольца Tm_M, находясь при этом в одной плоскости со связан н ы м ге п та зи нов ы м фрагментом ме л он а . Пл а н а рн ое ра с п оло же н и е этих фрагментов связано с эф ф е к т а ми с оп ря же н и я межд у д опантами и гептазиновым кольцом, которое по д к ре пле н о в озмо ж н ы м об ра зов а н и е м в о д ор од н ых с в язе й N…H–C. Электрон-донорные гетероциклы, замещающие по з ици и пр и вт о рич но м ат о м е азо т а м еж д у дв умя гепт аз ино вы ми кольцами, как правило, разверну т ы о р т о г о нал ь но по о т ношению к плоскости мелона. Молекулы мелона, допир о ванны е т ол ь ко электрон-акцепт о р ны ми зам ест ителями, содержащими в своей структуре хал ь ко г ены (O, S, Se), более планарны в случае Se как г ет ер о ат о м а в до пантах. Степень планарности мы определяли, как м аксим ал ь но е о т кл о нение ат о м о в, вх о д я щих в сост а в м ел о на, по ко о рдинат е Z от плоскости XY.
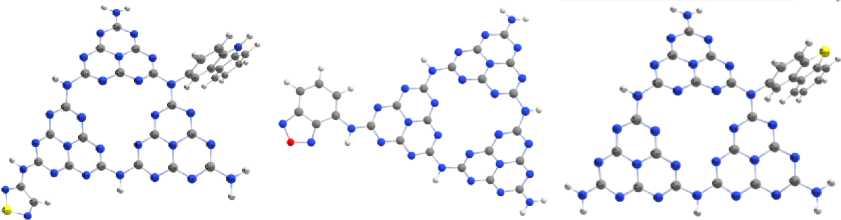
а) б) в)
Рис. 1. Примеры оптимизированных допированных структур мелона: а) Tm_S_D: акцептор – 1,2,5-тиадиазол, донор – карбазол, б) Tm_B_О: акцептор – 2,1,3-бензоксадиазол, в) Tm_D_S: донор – дибензотиофен
Например, для чистого Tm_M степень планарности составляет 2 , 37 Å. Молекулярное допирование чаще всего приводит к увеличению степени планарности в интервале 0 , 1–1 , 9 Å. Модели допированного мелона, построенные с учетом влияния растворителя, имеют более плоскую структуру, степень планарности уменьшается в среднем на 0 , 08 Å.
Рассмотрим изменение энергетических уровней ВЗМО и НСМО (рис. 2 и табл. 1) при модификации структуры мелона ковалентно-связанными допантами, а также разность граничных орбиталей (ΔE MO = Е НСМО – Е ВЗМО ). На рис. 2 видно, что модификация структуры любыми допантами закономерно влияет на изменение Е ВЗМО . Все значения энергий далее приведены относительно Е ВЗМО недопированного мелона, равной –6,32 эВ. Введение только электрон-донорных молекул повышает Е ВЗМО на 0,43…0,68 эВ, в среднем на 0,55 эВ. Одновременное присоединение к мелону электрон-донорных и электрон-акцепторных молекул приводит к повышению Е ВЗМО в среднем на 0,48 эВ. При введении только акцепторных молекул, больший вклад в изменение Е ВЗМО вносит атомная орбиталь более электроотрицательного атома. Поэтому для допантов, содержащих атом О, наблюдаемое изменение уровня ВЗМО сильнее, чем для соединений, содержащих атомы S и Se. Введение электрон-акцепторных молекул, представленных серией 1,2,5-халькогендиазолов, понижает значение Е ВЗМО на 0,15…0,24 эВ.
Рис. 2. Распределение значений энергий граничных орбиталей Е ВЗМО , Е НСМО в зависимости от состава и характера замещения атомов H в мелоне
Простой количественной зависимости изменения энергии НСМО от состава, положения и количества вводимых допантов не обнаружено (см. рис. 2). Максимальное понижение Е НСМО наблюдается для структуры, допированной только одним электрон-акцепторным допантом – 2,1,3-бензоселенадиазолом. Для структур, допированных дибензотиофеном и карбазолом, наблюдается незначительное повышение Е НСМО , на 0,05 эВ.
Анализ разности энергий граничных орбиталей ΔE MO показывает зависимость её значений от вида и количества вводимых допантов (рис. 3). Расчетное значение ΔEMO для мелона Tm_M составляет 3,57 эВ. Отметим, что близкий результат (3,88 эВ) был получен в теоретических расчетах, опубликованных ранее [48]. В целом величина разности энергий ВЗМО и НСМО уменьшалась при введении любых исследованных допантов. Максимальное уменьшение ΔEMO отмечается для структур, допированных одновременно и электрон-донорными, и электрон-акцепторными молекулами. Так, присутствие фрагментов 2,1,3-бензоселенадиазола и карбазола уменьшает ΔE MO на 1,04 эВ по сравнению с Tm_M. Введение только одного из этих допантов уменьшает ΔE MO максимально на 0,6 эВ.
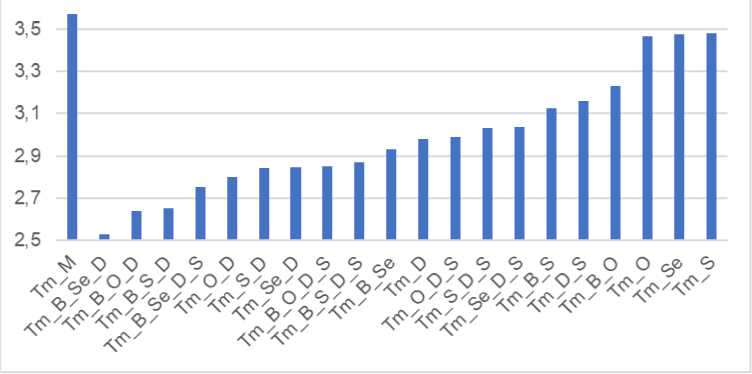
Рис. 3. Разность энергии граничных орбиталей ΔE MO , для структур с разными видами и количеством допантов
Анализ распределения электр онной плотности по молекулярным орбитал ям
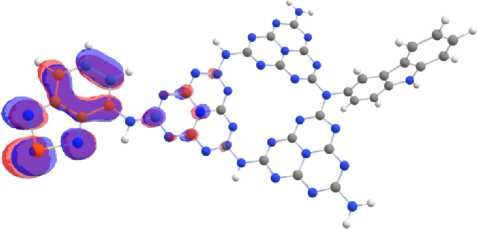
Ковалентное допирование м елона только акцептирующими электронну ю плотность бенз о- халькодиазолами приводит к смещению локализации ВЗМО в основном на акцептор (рис. 4). НСМО имеет большую площадь распределения, локализуясь одновременно и на фрагменте а к цептора, и на связанном с акцеп тором гептазиновом кольце. Допирование к арбазолом как эле к-трон- донорной молекулой приводит к локализации ВЗМ О на донорном фрагменте молекулы, при этом НСМО локализована на мелоне (рис. 5).
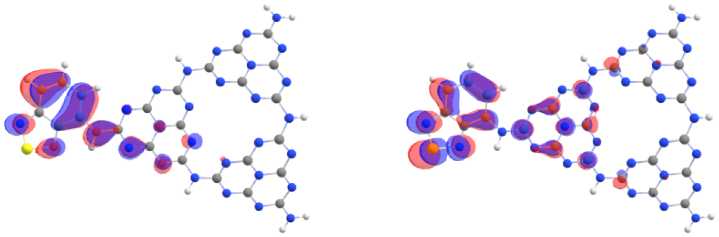
а)
б)
Рис. 4. Локализация ВЗМО (а) и НСМО (б) на молекуле мелона, допированной 2,1,3-бензотиадиазолом
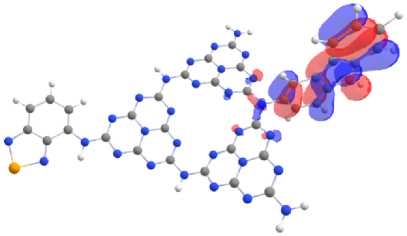
Одновременное допирование донорными и акцепторными молекулами, наиболее эффектив но уменьшающее разность грани чных молекулярных орбиталей, приводит к одинаковой закон омерности: ВЗМО оста ется локализованной на электрон -донорном фрагменте, а НСМО локализуется на электрон- акцепторном фрагменте и связанном с ним гептазиновом кольце (рис. 6). Говоря более конкретно, показано, что ВЗМО допированного мелона преимущест венно локализована на атоме N и распределена на связях С –С молекулы карбазола, а НСМО локализована на атомах N халькогенсодержащих эле ктрон -акцепторных допантов, связанных с гептазиновым фрагментом.
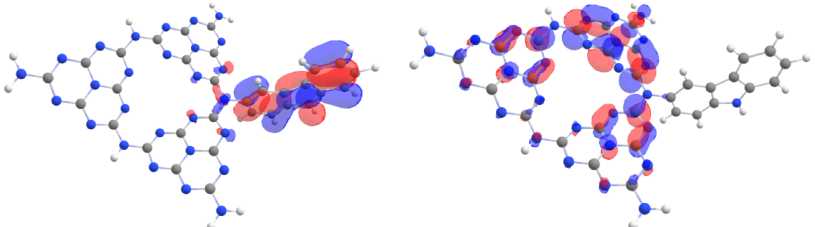
а)
б)
Рис. 5. Локализация ВЗМО (а) и НСМО (б) на молекуле мелона, допированной карбазолом
а) б)
Рис. 6. Локализация ВЗМО (а) и НСМО (б) на молекуле мелона, допированной молекулами 2,1,3-бензоселенадиазола и карбазола
В целом стоит отметить, что введение допантов в структуру мелона приводит не только к уменьшению ΔEMO, но и к изменению локализации ВЗМО и НСМО в структуре молекулы. Это может способствовать образованию сайтов связывания на поверхности допированной молекулы по отношению к наиболее способным к сорбции модельным молекулам. Так, были исследованы взаимодействия в комплексах, состоящих из бензилового спирта и мелона, которые являются переходными в реакциях окисления бензиловых спиртов до альдегидов с использованием фотокатализатора.
Нековалентные взаимодействия в комплексах ГНУ с молекулой бензилового спирта
Рассмотрим нековалентные взаимодействия в комплексах, образованных молекулами допи-рованного мелона и бензилового спирта ( табл . 3), и оценим прочность связывания в комплексе, используя топологические характеристики электронной плотности [55] в критических точках связи в зависимости от вида допанта. При моделировании наблюдается, что молекула бензилового спирта, как правило, ориентируется практически параллельно псевдоплоскости допированного мелона и формирует несколько связевых путей, определяемых нековалентными взаимодействиями (рис. 7). Значения электронной плотности в критических точках нековалентных связей показывают, что наиболее сильное взаимодействие наблюдается для водородной связи О–H…N–C, образованной атомом Н в молекуле спирта и вторичным атомом N в гептазиновом фрагменте.
Таблица 3
Смоделированные комплексы молекулярно-допированных структур мелона с бензиловым спиртом, энергии взаимодействия молекул E int и энергии водородных связей E НВ в соответствующих комплексах
|
№ |
Обозначение |
Структура |
Eint, ккал/моль |
EНВ, ккал/моль |
|
1 |
Tm_M |
–7,32 |
–3,92 |
|
|
2 |
Tm_B_Se |
–7,22 |
–3,84 |
|
|
3 |
Tm_D |
^^^^? |
–7,44 |
–3,94 |
Окончание табл. 3
|
№ |
Обозначение |
Структура |
E int , ккал/моль |
E НВ , ккал/моль |
|
4 |
Tm_B_Se_D |
V*frVv5»?r |
–7,22 |
–3,99 |
|
5 |
Tm_B_O |
^^^Vvy*^**^' |
–7,47 |
–3,87 |
|
6 |
Tm_B_S |
–7,37 |
–3,83 |
|
|
7 |
Tm_B_S_D |
^«*v^£у/ V- |
–7,43 |
–3,93 |
|
8 |
Tm_B_O_D |
–7,75 |
–3,98 |
Рис. 7. Формирование связевых путей в комплексе между бензиловым спиртом и молекулой допированного мелона
Энергия взаимодействия молекул в комплексе Eint сравнивалась с энергией водородной связи EНВ, определяемой исходя из корреляции, предложенной Эспиноза с соавт. в работе [52]. Определенное отсутствие количественной корреляции энергии взаимодействия в 8 исследованных комплексах бензилового спирта и допированного мелона с энергией водородной связи говорит о том, что не только одна водородная связь О–H…N–C определяет прочность связывания молекул бензилового спирта на поверхности допированного мелона в молекулярных комплексах. По- этому нами также проведен анализ зависимости энергии взаимодействия от ЕВЗМО допированного мелона. Для комплексов, допированных карбазолом (рис. 8), с ростом ЕВЗМО энергия взаимодействия Eint меняется сильнее по сравнению с комплексами, допированными только акцепторными заместителями. В последних при повышении ЕВЗМО на 0,33 эВ энергия взаимодействия повышается на 0,25 ккал/моль.
При допировании мелона карбазолом наблюдается иная тенденция: при незначительном увеличении Е ВЗМО – на 0,02 эВ – E int увеличивается на 0,53 ккал/моль. Селенсодержащие комплексы имеют меньшие значения E int , чем комплексы с S и O. Установленная зависимость косвенно указывает на то, что нитрид углерода в образуемом комплексе с бензиловым спиртом выступает донором электронной плотности. Отмечено, что формирование более прочных комплексов может препятствовать частичному окислению спиртов. В случае допирования мелона электрон-акцепторными селенсодержащими структурами образуется промежуточный комплекс с менее прочными связями, чем с серой и кислородом, и поэтому окисление проходит более эффективно.
-7,1
-7,2
-7,3
8 -7,4
-7,5
-7,6
-7,7
-7,8
Tm_B_Se Tm_B_Se_D
Tm_M
Tm_B_S
Tm_B_S_D Tm_D
Tm_B_O
Tm_B_O_D
-6,7 -6,6 -6,5 -6,4 -6,3 -6,2 -6,1 -6,0 -5,9 -5,8
ЕВЗМО, эВ
Рис. 8. Зависимость энергии взаимодействия молекул бензилового спирта и ГНУ E int в комплексе от Е ВЗМО мелона и его допированных производных
Характеристики одномерных периодических моделей допированного мелона
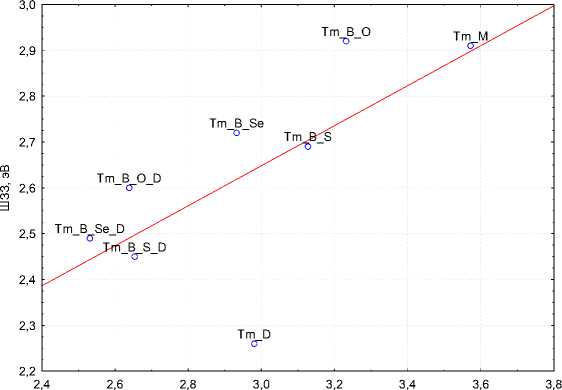
Далее мы рассмотрим 1D-полимерные модели допированного мелона, полученные с использованием периодических граничных условий, и сравним значения ширины запрещенной зоны, рассчитанные для этих полимерных структур, и величины разности энергий граничных орбиталей ΔE MO , полученные для молекулярных моделей (рис. 9, см. табл. 2). Из указанного сравнения можно оценить, насколько может быть корректен переход от газофазного кластерного расчёта к расчёту с периодическими граничными условиями и насколько полученные значения при этом переходе приближаются к экспериментальным. Значение ШЗЗ для немодифицированного полимерного мелона, равное 2,91 эВ, оказывается значительно ближе к экспериментальному (2,7 эВ, [54]), чем ΔE MO , равное 3,57 эВ, которое рассчитано для газофазного состояния. Здесь, однако, следует отметить, что расчёт равновесной геометрии полимерного мелона с необходимыми параметрами требует на порядки б о льших компьютерных ресурсов и времени, чем расчёт фрагмента мелона в газофазном состоянии.
Тем не менее, как видно из обобщающего рис. 9, прослеживается некоторая зависимость между значениями ШЗЗ и ΔE MO . Мелон, допированный кислородсодержащими гетероциклами, имеет большее значение ШЗЗ и ΔE MO . Селенсодержащие допанты, наоборот, максимально уменьшают значения ШЗЗ и ΔEMO. Однако корреляция этих значений в ряду меняющихся в допантах халькогенов (O, S, Se) низкая.
ΔE MO , эΒ
Рис. 9. Зависимость между значениями ШЗЗ и ΔE MO для исследуемых молекул
Из сравнения результатов расчётов в разных приближениях мы не можем сказать однозначно, какое допирование необходимо выполнить для максимального уменьшения ШЗЗ, так как при кластерном расчете наименьшую ΔE MO показывает мелон, допированный карбазолом и 2,1,3-бензоселенадиазолом. При периодических расчетах наименьшая ШЗЗ у мелона, допированного только электрон-донорной молекулой – карбазолом, затем следуют молекулы, допированные карбазолом и 2,1,3-бензотиадиазолом, карбазолом и 2,1,3-бензоселенадиазолом. Причем разница между ШЗЗ в двух последних случаях составляет 0,04 эВ. По рис. 9 также видно, что допирование мелона 2,1,3-бензоксадиазолом, по сравнению с другими допантами этого ряда, увеличивает ШЗЗ, даже по сравнению с недопированным мелоном.
Сравнение экспериментальных значений ШЗЗ и рассчитанных электронных свойств для ряда рассчитанных комплексов (табл. 4) показало следующее. При 5 %-ном допировании нитрида углерода бензо[c][1,2,5]халькогенадиазолами рассчитанные значения ШЗЗ и ΔE MO коррелируют с экспериментально измеренными значениями ШЗЗ 5% с коэффициентами 89 и 84 % соответственно (см. табл. 4). Изменение метода и уровня молекулярного расчета влияет на значения ΔE MO , при этом корреляция уменьшилась на 10 % при использовании метода учёта растворителя PCM и уровня HSE06/DZVP. Значения ШЗЗ и ΔE MO , полученные на уровне B3LYP/6-311+G(d,p), всегда имеют более высокое значение, чем для уровня HSE06/DZVP. Рассчитанное значение ШЗЗ для исходного ГНУ, равное 2,91 эВ, намного ближе к экспериментальным значениям 2,56 эВ [36] и 2,70 эВ [54], чем ΔE MO , рассчитанное для молекулы ГНУ в газофазном состоянии и равное 3,57 эВ.
Таблица 4
Экспериментальные и расчетные значения ШЗЗ и ΔE MO для исследуемых систем с электрон-акцепторными допантами
|
Обозначение |
ΔEMO, эВ |
EВЗМО, эВ |
Е НСМО , эВ |
ШЗЗ, эВ |
E(TVB*), эВ |
E(BVB*), эВ |
ШЗЗ 5% , эВ |
|
Tm_M |
3,57 |
–6,32 |
–2,75 |
2,91 |
–5,72 |
–2,81 |
2,56 (2,70) |
|
Tm_B_O |
3,23 |
–6,40 |
–3,16 |
2,92 |
–5,72 |
–2,80 |
2,41 |
|
Tm_B_S |
3,13 |
–6,23 |
–3,10 |
2,69 |
–5,65 |
–2,96 |
2,00 |
|
Tm_B_Se |
2,93 |
–6,13 |
–3,20 |
2,72 |
–5,65 |
–2,93 |
1,88 |
* – TVB – верхний валентный уровень, BVB – нижний виртуальный уровень.
Учитывая вышеизложенное, мы можем отметить, что результаты, полученные на обоих расчетных уровнях, приемлемо соответствуют экспериментальным данным, а именно тенденциям изменения ширины запрещенной зоны при допировании исходной структуры ГНУ электрон-акцепторными молекулами. В то же время, моделирование полимера с периодическими граничными условиями позволяет рассчитать эти значения и их тренды при допировании различными бензо[c][1,2,5]халькогенадиазолами несколько ближе к самим экспериментальным значениям и, возможно, правильно предсказать их для других электрон-акцепторных заместителей. С другой стороны, расчет равновесной геометрии полимера ГНУ требует на порядки больше компьютерных ресурсов и времени, чем расчет геометрии молекулы ГНУ в газофазном состоянии.
Заключение
Проведено моделирование структур мелона, допированного рядом гетероциклических молекул с выраженными электрон-донорными и электрон-акцепторными свойствами, в 2-х приближениях. Во-первых, мы применили подход, основанный на расчетах изолированных молекул. Во-вторых, мы использовали периодические граничные условия для моделирования 1D полимерных цепей. Сравнивая рассчитанные значения ШЗЗ и ΔEMO с экспериментальными данными для исходного и допированного ГНУ, мы пришли к выводу, что газофазные молекулярные расчеты больше подходят для качественной оценки трендов ΔE MO . Расчеты же полимерных цепей с периодическими граничными условиями дают удовлетворительные количественные зависимости ШЗЗ от вида и количества вводимого допанта. Одновременное допирование мелона электрон-донорными молекулами карбазола и электрон-акцепторными молекулами 2,1,3-бензохалькодиазолов максимально уменьшает разность энергий граничных орбиталей, а допирование только карбазолом максимально снижает ширину запрещенной зоны в рассмотренном ряду соединений.
Анализ распределения молекулярных орбиталей показал, что при эффективном допировании, максимально сужающем ΔE MO, ВЗМО локализовано на электрон-донорном фрагменте молекулы, а НСМО – на электрон-акцепторном фрагменте молекулы, что способствует быстрому рекомбинированию разделенных зарядов.
Допированные структуры ГНУ способны к образованию комплексов с бензиловым спиртом, образуемых за счет разных видов нековалентных взаимодействий. Селенсодержащие допанты ГНУ являются более подходящими молекулами для модификации катализатора в реакциях частичного окисления бензиловых спиртов.
Список литературы Ковалентное допирование нитрида углерода карбазолом и бензохалькодиазолами: моделирование электронных свойств в альтернативных приближениях
- Zou Z., Ye J., Sayama K. et al. // Nature. 2001. V. 414, No. 6864. P. 625. DOI: 10.1038/414625a.
- Kapilashrami M., Zhang Y, Liu Y.S. et al. // Chem. Rev. 2014. V. 114, No. 19. P. 9662. DOI: 10.1021/cr5000893.
- Lan Y, Lu Y, Ren Z. // Nano Energy. 2013. V. 2, No. 5. P. 1031. DOI: 10.1016/j.nanoen.2013.04.002.
- Wang H. Investigations into carbon nitrides and carbon nitride derivatives. Ludwig-MaximiliansUniversität München, 2013. 121 p.
- Keßler F.K. Structure and reactivity of s-triazine-based compounds in C/N/H chemistry. Ludwig-Maximilians-Universität München, 2019. 222 p.
- Jürgens B., Irran E, Senker J. et al. // J. Am. Chem. Soc. 2003. V. 125, No. 34. P. 10288. DOI: 10.1021/ja0357689.
- Y. Wang T.M. // J. Mater. Chem. C. 2016. V. 4. P. 6200. DOI: 10.1039/C6TC01860B.
- Knyazeva E.A., Rakitin O.A. // Russ. Chem. Rev. 2016. V. 85, No. 10. P. 1146. DOI: 10.1070/RCR4649.
- Zhang J., Zhang M., Lin S. et al. // J. Catal. 2014. V. 310. P. 24. DOI: 10.1016/j.jcat.2013.01.008.
- Schwab M.G., Hamburger M., FengX. et al. // Chem. Commun. 2010. V. 46, No. 47. P. 8932. DOI: 10.1039/C0CC04057F.
- Kailasam K., Schmidt J., Bildirir H. et al. // Macromol. Rapid Commun. 2013. V. 34, No. 12. P. 1008. DOI: 10.1002/marc.201300227.
- Yu Y, Yan W., Gao W. et al. // J. Mater. Chem. A. 2017. V. 5, No. 33. P. 17199. DOI: 10.1039/C7TA05744J.
- Zhang J., An X., Lin N. et al. // Carbon. 2016. V. 100. P. 450. DOI: 10.1016/j.carbon.2016.01.027.
- Cao J., Wu Q., Zhao Y. et al. // Appl. Catal., B. 2021. V. 285. P. 119817. DOI: 10.1016/j.apcatb.2020.119817.
- Shiraishi Y., Kanazawa S., Kofuji Y. et al. // Angew. Chem., Int. Ed. Engl. 2014. V. 53, No. 49. P. 13454. DOI: 10.1002/anie.201407938.
- Guo Y., Chu S., Yan S. // Chem. Commun. 2010. V. 46, No. 39. P. 7325. DOI: 10.1039/c0cc02355h.
- Chu S, Wang Y, Guo Y. et al. // ACS Catal. 2013. V. 3, No. 5. P. 912. DOI: 10.1021/cs4000624.
- Liu P., Sun N, Liang Y. et al. // Res. Chem. Interned. 2018. V. 44, No. 2. P. 843. DOI: 10.1007/s11164-017-3139-5.
- Chen Z, Sun P, Fan B. et al. // Appl. Catal., B. 2015. V. 170-171. P. 10. DOI: 10.1016/j.apcatb.2015.01.024.
- Fan X., Zhang L., Cheng R. et al. // ACS Catal. 2015. V. 5, No. 9. P. 5008. DOI: 10.1021/acscatal.5b01155.
- Ho W, Zhang Z., Lin W. et al. // ACS Appl. Mater. Interfaces. 2015. V. 7, No. 9. P. 5497. DOI: 10.1021/am509213x.
- Qin J., Wang S, Ren H. et al. // Appl. Catal., B. 2015. V. 179. P. 1. DOI: 10.1016/j.apcatb.2015.05.005.
- Zhang J., Chen X., Takanabe K. et al. // Angew. Chem., Int. Ed. Engl. 2010. V. 49, No. 2. P. 441. DOI: 10.1002/anie.200903886.
- Zheng Q., Durkin D.P., Elenewski J.E. et al. // Environ. Sci. Technol. 2016. V. 50, No. 23. P. 12938. DOI: 10.1021/acs.est.6b02579.
- Gao B., Wang J., Dou M. et al. // Sep. Purif. Technol. 2020. V. 241. P. 116576. DOI: 10.1016/j.seppur.2020.116576.
- Zheng D., Pang C., Liu Y. // Chem. Commun. 2015. V. 47. P. 9706. DOI: 10.1039/C5CC03143E.
- Chen Y., Zhang J., Zhang M. et al. // Chem. Sci. 2013. V. 4, No. 8. P. 3244. DOI: 10.1039/c3sc51203g.
- Zhang G., WangX. // J. Catal. 2013. V. 307. P. 246. DOI: 10.1016/j.jcat.2013.07.026.
- Kailasam K., Mesch M.B., Mohlmann L. et al. // Energy Technol. 2016. V. 4, No. 6. P. 744. DOI: 10.1002/ente.201500478.
- Li K., Zhang W. // Small. 2018. V. 14, No. 12. P. 1703599. DOI: 10.1002/smll.201703599.
- Fan X., Zhang L., Wang M. et al. // Appl. Catal., B. 2016. V. 182. P. 68. DOI: 10.1016/j.apcatb.2015.09.006.
- Zhang J., Zhang G., Chen X. et al. // Angew. Chem., Int. Ed. Engl. 2012. V. 51, No. 13. P. 3183. DOI: 10.1002/anie.201106656.
- Ye C., Li J.X., Li Z.J. et al. // ACS Catal. 2015. V. 5, No. 11. P. 6973. DOI: 10.1021/acscatal.5b02185.
- Zhang S., Li J., Wang X. et al. // J. Mater. Chem. A. 2015. V. 3, No. 18. P. 10119. DOI: 10.1039/C5TA00635J.
- Ong W.J., Tan L.L., Ng Y.H. et al. // Chem. Rev. 2016. V. 116, No. 12. P. 7159. DOI: 10.1021/acs.chemrev.6b00075.
- Chernukha A., Zirnik G., Mustafina K. et al. // Bulletin of the South Ural State University. Ser. Chemistry. 2022. V. 14, No. 4. P. 96. DOI: 10.14529/chem220410.
- Tyborski T., Merschjann C., Orthmann S. et al. // J. Phys.: Condens. Matter. 2013. V. 25, No. 39. P. 395402. DOI: 10.1088/0953-8984/25/39/395402.
- Thomas A., Fischer A., Goettmann F. et al. // J. Mater. Chem. 2008. V. 18, No. 41. P. 4893. DOI: 10.1039/b800274f.
- Zambon A., Mouesca J.M., Gheorghiu C. et al. // Chem. Sci. 2016. V. 7, No. 2. P. 945. DOI: 10.1039/C5SC02992A.
- Melissen S., Steinmann S.N., Le Bahers T. et al. // J. Phys. Chem. C. 2016. V. 120, No. 43. P. 24542. DOI: 10.1021/acs.jpcc.6b06335.
- Chen Z, Sun P, Fan B. et al. // Appl. Catal., B. 2015. V. 170-171. P. 10. DOI: 10.1016/j.apcatb.2015.01.024.
- Silva A.M., Rojas M.I. // Comput. Theor. Chem. 2016. V. 1098. P. 41. DOI: 10.1016/j.comptc.2016.11.004.
- Melissen S., Le Bahers T., Steinmann S.N. et al. // J. Phys. Chem. C. 2015. V. 119, No. 45. P. 25188. DOI: 10.1021/acs.jpcc.5b07059.
- Steinmann S.N., Melissen S.TAG, Le Bahers T. et al. // J. Mater. Chem. A. 2017. V. 5, No. 10. P. 5115. DOI: 10.1039/C6TA08939A.
- Frisch M.J., Trucks G.W., Schlegel H.B. et al. Gaussian 16 Revision C.01. 2016.
- Tomasi J, Mennucci B, Cammi R. // Chem. Rev. 2005. V. 105, No. 8. P. 2999. DOI: 10.1021/cr9904009.
- Dovesi R., Erba A, Orlando R. et al. // Wiley Interdiscip. Rev.: Comput. Mol. Sci. 2018. V. 8, No. 4. P. 1360. DOI: 10.1002/wcms.1360.
- Li K., Zhang W.D. // Small. 2018. V. 14, No. 12. P. 1703599. DOI: 10.1002/smll.201703599.
- Krukau A.V., Vydrov O.A., Izmaylov A.F. et al. // J. Chem. Phys. 2006. V. 125, No. 22. P. 224106. DOI: 10.1063/1.2404663.
- Vilela Oliveira D., Laun J., Peintinger M.F. et al. // J. Comput. Chem. 2019. V. 40, No. 27. P. 2364. DOI: 10.1002/jcc.26013.
- Balabin R.M. // J. Chem. Phys. 2008. V. 129, No. 16. P. 164101. DOI: 10.1063/1.2997349.
- Espinosa E, Molins E, Lecomte C. // Chem. Phys. Lett. 1998. V. 285, No. 3-4. P. 170. DOI: 10.1016/S0009-2614(98)00036-0.
- Chemcraft - graphical software for visualization of quantum chemistry computations. Version 1.8, build 654. https://www.chemcraftprog.com.
- Wang X., Maeda K., Thomas A. et al. // Nat. Mater. 2009. V. 8, No. 1. P. 76. DOI: 10.1038/nmat2317.
- Bader R.F.W. Atoms in Molecules: A Quantum Theory. Clarendon Press; 1990. 438 p.
