Квантово-химическое изучение целентеразина с учетом окружения и электронных корреляций

Автор: Антипина Любовь Юрьевна, Овчинников Сергей Геннадьевич
Журнал: Сибирский аэрокосмический журнал @vestnik-sibsau
Рубрика: Математика, механика, информатика
Статья в выпуске: 2 (35), 2011 года.
Бесплатный доступ
Электронная структура и полная энергия различных изомерных форм целентеразина рассчитаны методами квантовой химии, как в одноэлектронном приближении, так и с учетом корреляционных эффектов. Показано, что учет электронных корреляций позволяет выбрать структуру целентеразина CLZ(1H) как наиболее вероятную из возможных изомерных форм.
Обелин, квантово-химические расчеты, целентеразин, биолюминесценция, электронная корреляция
Короткий адрес: https://sciup.org/148176565
IDR: 148176565 | УДК: 538.958,
Quantum-chemical study of coelenterazine with the account of environment and electronic correlations
Electronic structure and total energy of various isomeric forms of coelenterazine has been calculated by methods of quantum chemistry both in one-electronic approach, and with correlation effects. It is shown that the account of electronic correlations allows choosing structure of coelenterazine CLZ (1H) as most probable of possible isomeric forms.
Текст научной статьи Квантово-химическое изучение целентеразина с учетом окружения и электронных корреляций
Явление биолюминесценции широко распространено в природе и лежит в основе целого ряда перспективных методов изучения биологических процессов. Са2+-регулируемые фотопротеины являются одними из наиболее изученных представителей биолю-минесцентных систем. Исследуются как сами белки, так и катализируемые ими реакции. Для многих белков идентифицированы субстраты реакций, определены их структуры и предложен механизм их окисления, приводящий к образованию электронно-возбужденных состояний [1–3]. Тем не менее до сих пор еще не решен один из основных фундаментальных вопросов в области биолюминесценции: каким образом белковая молекула препятствует диссипации возбужденных состояний в тепло, обеспечивая тем самым высокий квантовый выход хемилюминесцентной реакции. Восполнить этот пробел могут современные методы квантовой химии.
Для понимания физических процессов, происходящих в активном центре фотопротеина, в первую очередь необходимо определить реальную атомную структуру центральной молекулы целентеразина (CLZ) среди нескольких возможных изомерных форм. В данной работе эта задача решается с помощью микроскопических квантово-химических расчетов молекулы CLZ.
Методы квантовой химии, основанные на методе Хартри–Фока в качестве отправной точки и использующие представление о волновой функции как характеристике состояния квантовой системы, в принципе дают достаточно точный ответ о строении, энергии и химических свойствах исследуемого соединения. Для этого необходим полный учет энергии коррелированного движения электронов. В настоящий момент такие расчеты в ab initio представлении возможны для простых молекул, содержащим порядка 10…30 тяжелых атомов [4]. В связи с большим количеством атомов в системе применение первопринцип-ных методов ab initio видится затруднительным, так как занимает большое количество времени и компьютерных ресурсов. В данной работе использовались полуэмпирические методы РМ3 и РМ6 [5–8]. Большинство полуэмпирических методов параметризова- лось для органических молекул и биологических систем. На выбор метода в большей степени повлияло то, что полуэмпирические методы Хартри–Фока PM3 и РМ6 учитывают электростатическое взаимодействие, что очень важно в случае биологических систем. В данных методах все параметры, аппроксимирующие интегралы взаимодействия, подбираются наилучшим образом (оптимизируются с помощью набора соединений с надежно измеренными экспериментальными свойствами). Методы PM3 и РМ6 обеспечивают достаточную точность для воспроизведения многих физико-химических свойств молекул. Улучшение расчетов также достигалось путем применения метода СI (конфигурационное взаимодействие) для расчета основного и возбужденного состояния, что позволило учесть электронную корреляцию. Расчет проводился с использованием программы MOPAC2007.
Изомерные формы целентеразина. Целентеразин был выделен впервые в начале 1960-х гг. Структура была идентифицирована только в 1977 г. [9]. Немо-дифицированная форма CLZ отвечает формуле С 26 H 21 O 3 N 3 . Целентеразин может быть выкристаллизован из метанола как желто-оранжевые кристаллы. В метаноле CLZ флуоресцирует в желтой области и его ультрафиолетовый спектр поглощения имеет максимум на 435 нм [9, с. 470; 10]. В работе [11] проведено исследование немодифицированного CLZ и его аналогов в различных растворителях. На основании результатов этой работы было сделано предположение, что CLZ может существовать в нескольких изомерных формах: CLZ(2H), CLZ(7H) и CLZ(1H), протонированных соответственно в различных положениях (см. рисунок).
Форма CLZ(2H), протонированная в положение атома углерода С(2), имеет угол между остатком R1 и плоскостью имидазопиразиноных колец около 120°, в то время как у CLZ(1Н) и CLZ(7Н) угол составляет 180°, т. е. связь лежит в той же плоскости, что имида-зопиразиноные кольца. Таким образом, появление водорода при С(2) атоме приводит к изменению его конфигурации с sp2 на sp3, c последующим изменением длин связи и углов. При этом меняется и геометрия структуры, и заряды на атомах.
CLZ(7H)
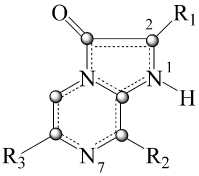
CLZ(1H)
Таутомерные формы целентеразина
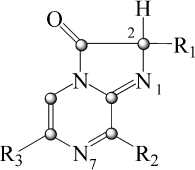
CLZ(2H)
Cравнение структур, рассчитанных в приближении Хартри–Фока и с учетом электронных корреляций. В связи с тем что формы CLZ(1H) и CLZ(7H) геометрически очень похожи, а рентгеноструктурный анализ не показывает положения атомов водорода в молекуле, определить, какая именно форма существует в белке или растворителях, экспериментально сложно. С другой стороны, для правильного определения спектральных характеристик необходимо знание точной атомной структуры молекулы. Даже небольшие изменения в атомной структуре приводят к заметным сдвигам в спектрах поглощения. В структуре целентеразина присутствуют атомы азота и кислорода с неподеленными электронными парами. Наличие данных атомов приводит к корреляционным эффектам, поэтому было решено проверить атомную структуру целентеразина с помощью одноэлектронных приближений и с учетом корреляционных поправок.
Энергии трех изомерных структур целентеразина, полученные различными методами, представлены в табл. 1. Энергия структуры CLZ(7H) принята за ноль, и остальные структуры сравнивались относительно нее.
При учете корреляции методом РМ6 уменьшается энергетическая разница между структурами CLZ(1Н) и CLZ(7Н). В случае РМ3 разница между этими структурами остается практически одинаковой.
Таблица 1
Энергия целентеразина, полученная различными методами, кДж/моль
|
Метод расчета |
Форма целентеразина |
||
|
CLZ(1H) |
CLZ(7H) |
CLZ(2H) |
|
|
РМ3 |
–4,70 |
0,00 |
–14,50 |
|
РМ3 CI |
–5,10 |
0,00 |
–14,91 |
|
РМ6 |
–17,58 |
0,00 |
–2,35 |
|
РМ6 CI |
–6,23 |
0 |
8,39 |
При этом следует отметить, что в любом случае структура CLZ(1H) получается более устойчивой. Но так как при учете электронной корреляции разница между двумя структурами небольшая, то логично предположить, что они могут переходить друг в друга в растворителях достаточно легко.
Также был проведен расчет данных структур в растворителях диметилсульфоксиде (DMSO) и метаноле (СН 3 ОН). Энергетические характеристики представлены в табл. 2.
Таблица 2
Энергия образования комплекса и расстояние до молекулы растворителя
|
Форма целенте-разина |
PM6 |
PM3 |
|
d E , кДж/моль (расстояние до растворителя, Å) |
d E , кДж/моль (расстояние до растворителя, Å) |
|
|
DMSO |
||
|
CLZ(1H) |
–126,98 (1,71) |
–90,89 (1,78) |
|
CLZ(7H) |
–10,92 (5,74) |
–20,84 (1,79) |
|
CLZ(2H) |
–45,71 (7,04) |
67,05 (2,48) |
|
СН 3 ОН |
||
|
CLZ(1H) |
–39,88 (1,75) |
–54,67 (2,45) |
|
CLZ(7H) |
–22,09 (1,86) |
–5,95 (2,74) |
|
CLZ(2H) |
–3,90 (2,11) |
–8,39 (2,77) |
Для учета влияния растворителя молекула целен-теразина была помещена в окружение из 10 молекул растворителя. Расчет проводился полуэмпирическими методами РМ3 и РМ6, чтобы учесть электростатическое взаимодействие. Было проведено сравнение устойчивости образующихся комплексов центральной молекулы с окружением растворителя. Расчет проводился следующим образом:
AE = E al, - X E , (1)
где A E - энергетический выигрыш при образовании комплекса, кДж/моль; E all – общая энергия, полученная из расчетов системы в целом, кДж/моль; E i – энергия каждого компонента системы, рассчитанная отдельно в вакууме, кДж/моль.
Также для оценки образования комплекса определялось расстояние между молекулой растворителя и водородом в положении N(1), N(7) и С(2) соответственно для каждого изомера в зависимости от структуры. Прямые методы показывают структуру CLZ(1H) устойчивей вне зависимости от растворителя.
В связи с тем что энергетически структуры отличаются не сильно, оценить, какая же именно структура реализуется в тех или иных условиях исходя только из энергетического фактора, нельзя. Поэтому было проведено сравнение длин связей для данных структур (табл. 3). Среднеквадратическое отклонение длин связей, полученных в результате оптимизации геометрии относительно эксперимента в вакууме и растворителях, определялось по формуле и =
X^
( n — i )
Таблица 3
Среднеквадратическое отклонение а длин связи для каждой структуры относительно эксперимента при расчете геометрии в растворителях, А
|
Метод расчета |
Растворитель |
Форма целентеразина |
||
|
CLZ(1H) |
CLZ(7H) |
CLZ(2H) |
||
|
PM3 |
Вакуум |
0,063 |
0,080 |
0,070 |
|
Вакуум с учетом корреляций методом CI |
0,059 |
0,081 |
0,070 |
|
|
DMSO |
0,059 |
0,070 |
0,074 |
|
|
CH 3 OH |
0,058 |
0,073 |
0,074 |
|
|
PM6 |
Вакуум |
0,062 |
0,071 |
0,072 |
|
Вакуум с учетом корреляций методом CI |
0,058 |
0,070 |
0,070 |
|
|
DMSO |
0,061 |
0,066 |
0,076 |
|
|
CH 3 OH |
0,058 |
0,064 |
0,074 |
|
Из представленных данных видно, что учет электронных корреляций приводит к заметному улучшению геометрии. Из сравнения геометрий видно, что наиболее близкая к экспериментальным данным структура – CLZ(1H). Все методы расчета показали отклонение для данной структуры меньше, чем для структуры CLZ(7Н) или CLZ(2H).
Исходя из всех факторов (геометрии, энергии, образования комплекса с растворителем), можно сделать вывод, что начальной структурой процесса активации молекулой кислорода будет являться структура CLZ(1H), а не CLZ(7H), как считалось ранее.
Спектральные характеристики изомерных форм целентеразина. В работе [11] было сделано предположение, что в различных растворителях це-лентеразин находится в различных изомерных формах. Апротонные растворители (DMSO и пр.) сдвигают спектр абсорбции в красную область (454 нм) в отличие от протонных растворителей (метанола) – 435 нм. Различные абсорбционные максимумы были приписаны различным таутомерным формам целенте-разина – протонированого в положении С(2) и N(7) соответственно.
Для проверки предположения Кормиера был выполнен расчет абсорбционных максимумов изомерных форм целентеразина в вакууме, метаноле и DMSO, используя процедуру CI на основе геометрий, полученных полуэмпирическим методом РМ3 CI с 10 занятыми и 6 свободными орбиталями.
Предварительно для оценки были проведены расчеты данных структур в вакууме (табл. 4). Полученные данные должны показать спектр абсорбции структур без влияния на них растворителей. Из данных, представленных в табл. 4, видно, что абсорбционный максимум структуры CLZ(2H), рассчитанный полуэмпирическим методом РМ3, показывает значение ~360 нм. Это означает, что мы можем исключить структуру CLZ(2H) из нашего рассмотрения при расчете полуэмпирическими методами, так как рассчитанный абсорбционный максимум очень сильно отличается от экспериментального.
При использовании процедуры CI в рамках метода РМ3 расчет показал сильный красный сдвиг для структуры CLZ(7H) при любых условиях. Таким образом, на основании полуэмпирических расчетов можно сделать следующее заключение: в апротонном растворителе CLZ существует в форме CLZ(7H), а в протонном – CLZ(1H), в отличие от того, что считалось ранее [11].
Таблица 4
Максимумы поглощения структур целентеразина в вакууме, нм
|
Форма целентеразина |
Вакуум |
DMSO |
CH 3 OH |
|
CLZ(1H) |
440 |
430 |
433 |
|
CLZ(7H) |
475 |
540 |
499 |
|
CLZ(2H) |
363 |
369 |
362 |
Итак, проведены расчеты различных изомерных форм целентеразина методами одноэлектронного приближения и с учетом электронных корреляций. Показано, что учет электронных корреляций дает структуру, более близкую к экспериментальной, и позволяет выбрать форму целентеразина CLZ(1H) как наиболее вероятную из возможных изомерных форм. Целентеразин в протонных растворителях находится в изомерной форме CLZ(1Н), в апротонных – в форме CLZ(7Н). Образования формы CLZ(2H) в растворителях не происходит. Изомерная форма CLZ(2H) может образовываться только при захвате целентеразина белковым окружением.
