Метилирование генов Р53-респонзивных онкосупрессорных микроРНК при гемобластозах

Автор: Воропаева Е.Н., Поспелова Т.И., Березина О.В., Чуркина М.И., Гуражева А.А., Максимов В.Н.
Журнал: Сибирский онкологический журнал @siboncoj
Рубрика: Обзоры
Статья в выпуске: 2 т.21, 2022 года.
Бесплатный доступ
Цель исследования - представить современные данные о частоте и значении метилирования генов ряда р53-респонзивных онкосупрессорных микроРНК при опухолевых заболеваниях системы крови. Материал и методы. Проведен поиск доступных литературных источников, опубликованных в базах данных pubMed и РИНЦ. Найдено 399 статей, из которых 62 были включены в данный обзор. Результаты. Белок р53 регулирует целый класс микроРНК - высококонсервативных малых молекул РНК, которые влияют на экспрессию генов в основном путем подавления трансляции. МикроРНК играют важную роль во всех клеточных процессах и могут иметь как онкосупрессорные, так и проонкогенные свойства. Нарушения экспрессии активируемых р53 онкосупрессорных микроРНК в различных опухолях могут быть связаны со специфическими эпигенетическими механизмами (метилированием ДНК и деацетилированием гистонов). В обзоре рассмотрены молекулярно-генетические характеристики онкосупрессорных микроРНК, функционирующих при нормальном кроветворении, нарушение экспрессии которых показано при развитии гемобластозов, а именно: miR-34a, miR-34b/с, miR-145, miR-143 и miR-203. Известно, что транскрипция генов этих микроРНК осуществляется и регулируется с собственных промоторов. Приведены последние опубликованные результаты исследований по диагностическому, прогностическому и клиническому значению метилирования генов рассматриваемых микроРНК при злокачественных новообразованиях системы крови. Согласно данным литературных источников, частыми общими мишенями для микроРНК miR-34a, miR-34b/с, miR-145, miR-143 и miR-203 являются м-РНК ряда проонокогенов, а именно: транскрипционного фактора C-MYC, позитивных регуляторов клеточного цикла в контрольной точке перехода G1/S фаз CDK4, CDK6 и CYCLIN-D1, антиапоптотических белков MDM2, MDM4, ВCL2 и MCL1, а также ДНК-метилтрансфераз DNMT3A и DNMT3B и других молекул. Описано наличие положительных обратных связей между р53 и активируемыми им микроРНК, а также отрицательных обратных связей между р53-респонзивными микроРНК и C-MYC и ДНК-метилтрансферазами. Заключение. Данные, представленные в обзоре, уточняют современные представления о работе регуляторной сети белка р53 и активируемых им микроРНК, а также подчеркивают функциональную ассоциацию р53-респонзивных микроРНК.
Тр53, р53, микрорнк, mir-34a, mir-34b/с, mir-145, mir-143, mir-203, гемобластозы, экспрессия, метилирование
Короткий адрес: https://sciup.org/140293893
IDR: 140293893 | УДК: 616.15-006:575.113:577.215.3
Methylation of P53-responsive oncosuppressive microRNA genes in hemoblastosis
The purpose of the study was to present up-to-date data on the frequency and significance of a number of p53-responsive oncosuppressive microRNAs genes methylation in malignant neoplasms of the blood system. Material and Methods. The search for available literary sources published in the PubMed and RISC databases was carried out. A total of 399 articles were found, of which 62 were included in this review. Results. The p53 protein regulates a whole class of microRNAs - highly conserved small RNA molecules that affect gene expression mainly by suppressing translation. МicroRNAs play an important role in all cellular processes and can have both oncosuppressive and pro-oncogenic properties. Impaired expression of p53-activated oncosuppressive microRNAs in various tumors may be associated with specific epigenetic mechanisms (DNA methylation and histone deacetylation). The review examines the molecular and genetic characteristics of oncosuppressive microRNAs functioning in normal hematopoiesis, the violation of expression of which is shown in the development of hemoblastoses, namely: miR-34a, miR-34b/c, miR-145, miR-143 and miR-203. It is known that the transcription of the genes of these microRNAs is carried out and regulated from their own promoters. The latest published research results on the diagnostic, prognostic and clinical significance of gene methylation of the microRNAs under consideration in malignant neoplasms of the blood system are presented. According to literature data, common targets for miR-34a, miR-34b/c, miR-145, miR-143 and miR-203 microRNAs are mRNAs of a number of pro-oncogenes, namely: transcription factor C-MYC, positive cell cycle regulators at the G1/S transition point of CDK4, CDK6 and CYCLIN-D1 phases, anti-apoptotic proteins MDM2, MDM4, BCL2 and MCL1, as well as DNMT3A and DNMT3B methyltransferases and other molecules. In this regard, it should be noted that there are positive feedbacks between p53 and microRNAs activated by it, as well as negative feedbacks between p53-responsive microRNAs and C-MYC and DNA methyltransferases. Conclusion. Thus, the data presented in the review clarify the current understanding of the work of the regulatory network of the p53 protein and the microRNAs activated by it, and also emphasize the functional association of p53-responsive microRNAs.
Текст научной статьи Метилирование генов Р53-респонзивных онкосупрессорных микроРНК при гемобластозах
Роль микроРНК в развитии опухолей в последние десятилетия активно изучается, поскольку они участвуют практически во всех клеточных процессах, модулирующих злокачественную трансформацию клеток, а именно: контроле клеточного цикла, ответе на повреждение ДНК, дифференцировке, пролиферации, апоптозе, старении, обмене веществ, эпителиально-мезенхимальном переходе и метастазировании [1]. Получены данные, что микроРНК действуют не только как внутриклеточные мессенджеры, но также могут секретироваться и циркулировать в крови как часть апоптотических тел, микровезикул и экзосом [2].
МикроРНК могут быть отнесены к классу онкосупрессоров или проонкогенов, хотя подобное разделение подходит не для всех молекул [3]. Уровень экспрессии первых в процессе неопластической трансформации клетки может снижаться, уровень вторых – повышаться. Хорошо изучены онкосупрессорные микроРНК кластера miR-34. Примером микроРНК с онкогенным потенциалом являются молекулы кластера miR-17-92. Входящие в него молекулы запускают пролиферацию, ангиогенез, эндотелиально-мезенхимальный переход и метастазирование при широком спектре опухолей [4]. Исследования по профилированию экспрессии генов и биоинформационный анализ показывают, что дерегуляция микроРНК является распространенным событием при новообразованиях. Опухоль имеет свой, отличный от нормальных клеток спектр экспрессии данных молекул.
Известно, что точная регуляция содержания микроРНК в гемопоэтических клетках необходима для нормального кроветворения. Так, существуют белки, участвующие в гемопоэзе, экспрессия которых должна очень четко регулироваться в клетке с помощью микроРНК, а ее нарушение приводит к различным патологическим состояниям, в т. ч. развитию злокачественных новообразований.
МикроРНК имеют важнейшую регуляторную функцию и в работе иммунной системы, начиная от поддержания пула стволовых клеток и заканчивая созреванием и функционированием Т- и В-лимфоцитов. Они не только влияют на направление развития клетки, но также могут играть более тонкую роль в придании клетке устойчивости или чувствительности к важнейшим биологическим процессам – апоптозу, пролиферации, дифференцировке и т.д.
Участие микроРНК в лимфомогенезе подтверждено в моделях на трансгенных мышах. Аберрантная экспрессия микроРНК не только приводит к развитию лимфопролиферативных заболеваний, но и способствует увеличению темпов их опухолевой прогрессии. Впервые значение дерегуляции микроРНК в биологии гемобластозов получило оценку, когда было показано, что в локусе 13q14, который часто подвергается делеции при хроническом лимфолейкозе, расположены гены онкосу-прессорных микроРНК miR-15a и miR-16-1 [5].
Аберрантная экспрессия микроРНК наблюдается и при лимфомах, например при диффузной В-клеточной крупноклеточной лимфоме (ДВККЛ). Показана возможность разделения ДВККЛ на классификационные подгруппы герминального и негерминального происхождения на основе спектра экспрессии микроРНК, отмечены различия в экспрессии микроРНК в клетках лимфомы и нормальной лимфоидной ткани [6].
Ряд микроРНК имеют однонаправленные изменения экспрессии при различных подтипах лимфом, таковым, например, является повышение экспрессии онкогенных miR-155, miR-17-92b и miR-21, а также снижение экспрессии онкосупрес-сорных miR-15а/16, miR-34а, miR-150 и miR-29 [3, 7].
Метилирование генов микроРНК
Изменения уровня микроРНК в клетках могут быть вызваны различными механизмами. Гиперэкспрессия их может быть результатом амплификации кодирующего микроРНК региона генома, промоции транскрипции или потери эпигенетического сайленсига гена микроРНК. Снижение уровня экспрессии, напротив, может быть связано с делецией участка хромосомы, на котором расположен ген микроРНК, наличием мутаций в предшественниках и ключевых последовательностях зрелых молекул, а также гиперметилированием промотора генов микроРНК.
Нарушение гомеостаза метилирования ДНК – центральное звено в эволюции опухолей. Основной чертой генома злокачественной клетки является его глобальное деметилирование, приводящее к генетической нестабильности. С другой стороны, при опухолях происходит аномальное гипермети- лирование ДНК в промоторных регионах генов-онкосупрессоров [8]. Причем такое аберрантное метилирование является ранним событием в эволюции опухолей и способствует дальнейшему прогрессированию заболевания [9].
Многие гены микроРНК находятся в интронах белок-кодирующих генов, и, следовательно, экспрессия микроРНК зависит от уровня транскрипции гена-«хозяина». Однако часть из вну-тригенных, а также межгенно расположенных микроРНК имеют свои собственные промоторы. Поскольку большинство кодирующих микроРНК генов обнаружено в CpG-богатых регионах, было высказано предположение, что метилирование ДНК может иметь важное значение в нарушении их транскрипции [4]. Позднее было показано, что промоторы генов микроРНК подвергаются метилированию в пять-десять раз чаще, чем генов, кодирующих белки, а их аберрантное гиперметилирование – один из наиболее значимых механизмов снижения экспрессии микроРНК при опухолях [9].
В настоящее время выявлено около 200 генов микроРНК, которые регулируются путем метилирования ДНК при множестве типов неоплазий. Было показано, что 45,5 % этих генов аберрантно метилированы, по меньшей мере, при двух типах онкологических заболеваний. Так, например, гиперметилирование генов MIR-34B, MIR-34C и MIR-34A было показано при 24, 21 и 17 видах рака соответственно. Снижение экспрессии он-косупрессорных микроРНК, например miR-203, miR-124-1 и miR-129-2, при злокачественных новообразованиях, в т. ч. системы крови, также происходит путем аберрантного метилирования. Ряд микроРНК считаются специфично метилированными при отдельных типах рака, однако предполагается, что этот феномен отчасти связан с малой изученностью вопроса и может измениться при исследовании метилирования этих генов при расширенном спектре опухолей [10].
Можно предположить, что аберрантное метилирование генов микроРНК при опухолях – потенциально полезный молекулярный биомаркер как в диагностике, так и в предикции, прогнозе опухоли и разработке стратегии таргетной терапии злокачественных новообразований.
МикроРНК как посредники эффектов р53
В физиологических условиях точная и согласованная регуляция р53-индуцируемых микроРНК опосредует его противоопухолевые эффекты. Так, реализация р53-опосредованного ответа на генотоксический клеточный стресс во многом осуществляется за счет изменения спектра экспрессируемых микроРНК, а именно активации онкосу-прессорных и подавления онкогенных молекул, что возможно как напрямую через транскрипционнозависимые механизмы, так и косвенно (рис. 1).
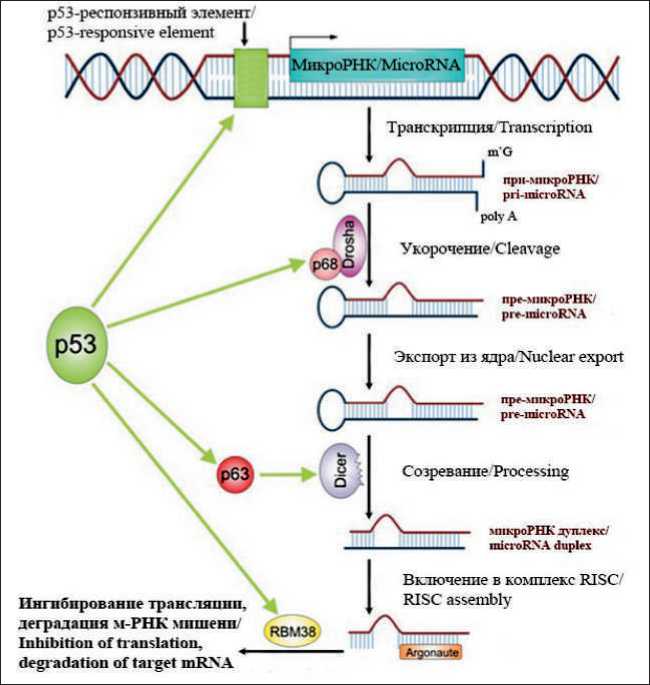
Рис. 1. Регуляция экспрессии микроРНК белком р53 на уровне транскрипции и созревания микроРНК (источник с изменениями [61]).
Fig. 1. Regulation of microRNA expression by p53 protein at the level of transcription and microRNA maturation.
Важно отметить, что вариабельность сигнатуры экспрессии р53-регулируемых микроРНК и их генов-мишеней зависит от типа клеток, природы стресса и причины повреждения ДНК. При этом регулируемые р53 микроРНК вовлечены в комплекс обратных и прямых связей с р53, что способствует совместному контролю, усилению и настройке сигналов в ответ на стресс.
Так, в генах ряда онкосупрессорных микроРНК, например, let-7, miR-16-2, miR-29b, miR-34a, miR-34b/c, miR-107, miR-145 и других, были выявлены р53-респонзивные элементы, связываясь с которыми, р53 индуцирует их транскрипцию. Экспрессия других микроРНК, таких как miR-107, miR-122, miR-182, miR-200, miR-205, miR-605 и miR-1204, также потенциально может регулироваться за счет прямого связывания р53 с промоторами генов, однако необходимы экспериментальные подтверждения такого взаимодействия [5].
Помимо прямой регуляции транскрипции возможно участие р53 в регуляции экспрессии микроРНК на посттранскипционном уровне путем влияния на процессинг данных молекул. Показано, что р53 усиливает посттранскрипционный процессинг онкосупрессорных микроРНК miR-16-1, miR-143 и miR-145 в ответ на повреждение ДНК как на уровне связывания с белковым комплексом Drosha, так и на уровне связывания с белковым комплексом Dicer [11, 12].
Большинство мутаций в гене ТР53 , ответственном за синтез р53, при злокачественных новообразованиях человека находятся в ДНК-связывающем домене, который имеет ряд функций [13]. Определение данных мутаций имеет большое значение для определения прогноза и тактики ведения больных НХЗЛ [14]. Давно известно, что ДНК-связывающий домен белка р53 не только контактирует с ДНК и позволяет активировать гены-мишени (включая гены микроРНК) для индуцирования апоптоза или остановки клеточного цикла, но и стимулирует запуск программируемой клеточной смерти через взаимодействие с белками семейства BCL2 в митохондриях. В экспериментах на мышах были получены убедительные доказательства того, что некоторые мутантные варианты р53 не просто утрачивают нормальные онкосупрессорные функции белка, но и приобретают стимулирующие опухолевый рост свойства, например, путем изменения спектра р53-регулируемых генов [15].
Позднее появились данные о том, что транскрипционно-неактивные мутантные варианты р53 препятствуют функциональной сборке комплекса Drosha, имеющего основную роль в инициации процессинга микроРНК в ядре. В частности, мутации в «горячих» точках р.R175H и р.R273H коррелируют с более низкой активностью Drosha, в то время как Р53, с мутацией р.C135Y, которая редко встречается при опухолях, не име- ет такого влияния на Drosha [16]. Эти данные свидетельствуют о том, что транскрипционнонезависимая модуляция биогенеза микроРНК является еще одним из аспектов противоопухолевой активности р53.
В последнее десятилетие возрос интерес к изучению метилирования генов микроРНК при гемобластозах. Активно изучается значение данных молекул в нормальной дифференцировке и функционировании кроветворных клеток и их дерегуляции в развитии опухолей. Несмотря на то, что общие знания о роли микроРНК в развитии злокачественных новообразований системы крови накапливаются быстрыми темпами [6, 17], данные по частоте и спектру метилирования генов микроРНК, участию метилирования онкосупрес-сорных микроРНК в прогрессии опухоли, его прогностической, предиктивной и диагностической ценности при гемобластозах немногочисленны.
Далее будут описаны имеющиеся данные о роли р53-респонзивных онкосупрессорных микроРНК и частоте метилирования кодируемых ими генов при злокачественных новообразованиях системы крови.
Метилирование генов р53-респонзивных микроРНК при опухолях системы крови Гены кластера miR-143/145
Гены MIR-143 и MIR-145 находятся на расстоянии 1.3 kb (килобаз) друг от друга в хромосомном регионе 5q33. Предполагается, что miR-143 и miR-145 образуются с общего предшественника и их функции кооперированы [18]. Геном-«хозяином» данного кластера микроРНК является ген некодирующей РНК CARMN . Известно, что нокдаун CARMN снижает экспрессию miR-143 и miR-145. С другой стороны, для кластера идентифицированы независимые транскрипционные факторы и отдельная промоторная регуляция экспрессии miR-145 [19].
Показано, что уровень miR-145 повышается в ответ на стресс через PI3K/Akt и p53-опосредованные пути. Транскрипция ее индуцируется при связывании р53 с соответствующим регуляторным элементом в промоторе гена [18]. В отличие от такого прямого воздействия участие белка р53 в регуляции экспрессии miR-143 происходит на посттранскрипционном уровне. Созревание pri-miR-143 в pre-miR-143 в ядре клетке запускается за счет связи p53 с компонентами комплекса Drosha [20].
Учитывая количество и спектр целевых генов, кластер miR-143/145 является важным для онкогенеза (табл. 1). Мишенями miR-145 являются онкоген BCL2, промоторы клеточного цикла ци-клинкиназы CDK4 и CDK6, циклин D1, регулятор деградации р53 убиквитинлигаза MDM2, а также транскрипционные факторы OCT4, SOX2 и KLF4, ответственные за самообновление недифференцированных эмбриональных стволовых клеток [18,
-
21] . Соответственно, выключение этих генов на посттрансляционном уровне за счет микроРНК приводит к активации апоптоза, снижению способности к пролиферации и подавлению плюрипотентности эмбриональных стволовых клеток.
Наиболее примечательное открытие в отношении miR-145 касается определения роли miR-145 в посттранскрипционной регуляции C-MYC . Так, miR-145 непосредственно нацелена на онкоген C-MYC [18]. Более поздние исследования показали, что мишенью данной микроРНК является также ген ERK5 . Кодируемый им белок участвует в передаче сигнала по MAPK-путям в ответ на стимулы факторов роста, полученные клеткой через рецепторные тирозинкиназы или рецепторы, сопряженные с G-белками, что в конечном итоге приводит к активации C-MYC [22]. Другими мишенями miR-145 являются ДНК метилтрансфераза DNMT3b и онкоген RAS . Предполагается, что между miR-145 и DNMT3b, К-RAS и С-MYC, а также miR-143 и DNMT3а, К-RAS существует перекрестная регуляция через двойные отрицательные контуры обратных связей [18, 22].
Биоинформационный анализ и исследования на культурах опухолевых клеток показывают, что потенциальными мишенями для miR-143 и miR-145 могут быть и ряд других м-РНК, в т. ч. транскрипционных факторов, обладающих онкогенными функциями [23], но необходимо экспериментальное подтверждение полученных таким образом данных для разных патологических состояний и типов тканей. Однако уже сейчас понятно, что низкая экспрессия miR-143 и miR-145 может привести к несбалансированному сигнальному каскаду, включающему MAPK, и устойчивой клеточной пролиферации, а также напрямую и косвенно приводит к усилению клеточного роста и выживания, подавлению апоптоза и увеличению способности к адгезии, инвазии и миграции клеток [24, 25].
В отличие от здоровых тканей экспрессия данных молекул снижена при опухолях. На материале, полученном от здоровых доноров, показано, что miR-143 высоко экспрессируется в гемопоэтических клетках костного мозга различной стадии дифференцировки. MiR-145, напротив, в здоровых гемопоэтических клетках костного мозга практически не обнаруживается [26]. Известно также, что miR-145 менее выраженно экспрессируется в В-клетках памяти в сравнении с наивными В-клетками [27]. Предполагается, что уровень miR-143 и miR-145 может служить биологическим маркером для отличия опухолевых и нормальных В-лимфоцитов. При В-клеточных лимфоидных новообразованиях экспрессия miR-145 и miR-143 резко снижена [28]. Даунрегуляция miR-145 была показана при ДВККЛ, в т. ч. при первичной ДВККЛ центральной нервной системы [29, 30].
В экспериментах на линиях клеток лимфомы Беркитта (ЛБ) также продемонстрировано снижение уровня miR-143 и miR-145 [28]. Восстановление экспрессии данных микроРНК при ЛБ, множественной миеломе (ММ) и Т-клеточном остром лимфобластном лейкозе (Т-ОЛЛ) приводило к снижению роста опухолевых клеток [31]. Кроме того, как в клеточных моделях in vitro, так и в мышиной модели ксенотрансплантата для множественной миеломы человека нормализация экспрессии miR-145 потенциировала противоопухолевую активность бортезомиба [32], а при раках обусловливала радиочувствительность опухоли [33].
На большом числе солидных новообразований (глиома, рак простаты, толстой кишки, яичников, легких, молочных желез, нейроэндокринные опухоли гипофиза) показано, что низкая экспрессия микроРНК кластера miR-143/145 зачастую связана с аберрантным метилированием [33, 34], а обработка культур опухолевых клеток деметилирующими агентами приводила к восстановлению их транскрипции [24, 34].
В отличие от злокачественных новообразований эпителиальной природы данных о статусе метилирования генов микро-РНК кластера miR-143/145 при гемобластозах и, в частности, при лимфомах практически нет. Единственное встреченное нами в литературных источниках сообщение об эпигенетическом выключении их экспрессии касается линии NK/T-клеточной лимфомы miR-143 [22].
Гены семейства miR-34
Семейство miR-34 включает в себя три онко-супрессорных микроРНК: miR-34a, miR-34b и miR-34c, для которых характерна высокая степень (более 80 %) гомологичности последовательности. Однако точная гомология соблюдается в районе их 5'-концов – районе расположения ключевой последовательности, необходимой для связывания с м-РНК мишенями. Геном-«хозяином» MIR-34A является EF570048 , а MIR-34B/C – ген BC021736 . При этом показано, что регуляция транскрипции MIR-34b/c может осуществляться и с промотора не только хозяйского гена, но и промотора противоположно ориентированного гена BTG4 . Промотор и сайт начала транскрипции MIR-34A был картирован более чем на 30 kb выше последовательности, кодирующей зрелую микроРНК [35].
Ген MIR-34A находится на 1-й хромосоме в регионе 1p36.22, тогда как ген MIR-34B/C – на хромосоме 11 в регионе 11q23.1. Последний отвечает за последовательность бицистронного транскрипта, с которого в последующем образуются две зрелые микроРНК. MIR-34A и MIR-34B/C были первыми генами микроРНК, для которых была показана прямая индукция транскрипции белком р53. Кроме того, miR34A может создавать положительную петлю обратной связи с р53 путем подавления деацетилазы SIRT1, что приводит к ацетилированию и активации р53. Даунрегуляция их экспрессии осуществляется белком C-MYC [36].
С помощью биоинформационного анализа и в экспериментальных исследованиях показано, что микроРНК семейства miR-34 имеют большое число общих мишеней, а именно, м-РНК регуляторов клеточного цикла в контрольной точке перехода G1/S фаз, репарации повреждений ДНК и программированной клеточной смерти, эпителиально-мезенхимального перехода, миграции, противоопухолевого имунного ответа и неоангиогенеза [37]. Соответственно, уменьшение экспрессии miR-34a и miR-34b/c способствует злокачественному росту и может наблюдаться в различных опухолях (табл. 1).
Метилирование промоторных областей генов семейства miR-34 является причиной снижения их экспрессии при широком круге злокачественных новообразований, например, при раке толстого кишечника, поджелудочной железы, молочных желез, яичников, почек и саркомах мягких тканей и других [38]. Промоторы генов MIR-34A и MIR-34B/C содержат CpG-островки, гиперметилирование которых приводит к нарушению транскрипции кодируемых ими микроРНК. Другой причиной снижения их экспрессии при злокачественных новообразованиях могут быть хромосомные делеции [39].
Изучение роли метилирования генов кластера miR-34 при опухолях системы крови выявило следующее. В образцах опухолевой ткани пациентов с впервые выявленной ДВККЛ и в клеточных линиях ДВККЛ метилирование MIR-34A и MIR-34B/C было обнаружено в 28 и 78 % случаев соответственно с высокой степенью корреляции между генами (р=0,001). При этом в нормальных Т- и В-клетках и ткани реактивных лимфоузлов метилирования генов микроРНК семейства miR-34 не выявлено [36].
При множественной миеломе нарушение экспрессии микроРНК кластера miR-34 также зачастую связано с аберрантным метилированием
Таблица 1/table 1
Краткие сведения об описанных р53-респонзивных онкосупрессорных микроРНКBrief information about the described p53-responsive oncosuppressive microRNas
|
Снижение |
Метилирование |
|||||
|
МикроРНК/ MicroRNA |
Ген/ Gene |
Расположение/ Location |
Локализация/ Localization |
Биологический процесс/ Biological process |
экспрессии микроРНК/ Reduction of |
гена микроРНК/ Methylation of the microRNA |
|
microRNA |
||||||
|
gene |
||||||
|
expression |
||||||
|
ПЛЦНС, Т-ОЛЛ, ДВККЛ, ЛМЗС, Нет анных/ miR-145 Контроль клеточного цикла, В-НХЛ, ЛБ/ е N т o да d н a н ta ых апоптоз, дифференцировка PLCNS, T-ALL, нутригенное, клеток, клеточная DLBCL, LMZS, ген-«хозяин» адгезия, эпителиально- B-NHL, BL MIR- CARMN/ 5q33 мезенхимальный переход/ ОЛЛ, В-НХЛ, 143/145 Intragenic, Cell cycle control, apoptosis, , ЛБ , host gene , CARMN cell differentiation, cell adhe- NK/T-клеточная NK/T-клеточная sion, epithelial-mesenchymal лимфома/ лимфома/ miR-143 transition ALL, B-NHL, NK/T-cell lym- HL, phoma NK/T-cell lymphoma |
|
Контроль клеточного ДВККЛ, Внутригенное, цикла, репарация ДНК, ХЛЛ, ДВККЛ, NK/T-клеточная ген-«хозяин» MIR- апоптоз, эпителиально- ЛБ, ЛХ/ лимфома, ЛХ/ miR-34а EF570048/ 1p36.22 34A мезенхимальный переход, CLL, DLBCL, DLBCL, NK/T- Intragenic, host противоопухолевый BL, HL cell lymphoma, gene EF570048 иммунный ответ, HL неоангиогенез/ нутригенное, Cell cycle control, DNA ген-«хозяин» ХЛЛ, ММ, MIR- repair, apoptosis, epithelial- ХЛЛ, ДВККЛ/ miR-34b/c 34B/C BC021736/ 11q23.1 mesenchymal transition, ДВККЛ/CLL, CLL, DLBCL Intragenic, host MM, DLBCL antitumor immune response, gene BC021736 neoangiogenesis |
|
Дифференцировка клеток, пролиферация и клеточный рост, апоптоз, репарация Т-НХЛ, ОМЛ, ДНК, радиационная ХЛЛ, В-НХЛ, miR-203 MIR-203 Межгенное/ 14q32.33 чувствительность, ММ, ХМЛ, ЛХ/ ММ, ХМЛ, ЛХ/ Intergenic неоангиогенез/ MM, CML, HL T-NHL, MM, Cell differentiation, prolifera- CLL, HL tion and cell growth, apop tosis, DNA repair, radiation sensitivity, neoangiogenesis |
Примечание: ПЛЦНС – первичная лимфома центральной нервной системы; ОЛЛ – острый лимфобластный лейкоз; ОМЛ – острый миелобластный лейкоз; ДВККЛ – диффузная В-клеточная крупноклеточная лимфома; ЛМЗС – лимфома маргинальной зоны селезенки; В-НХЛ – В-клеточные неходжкинские лимфомы; T-НХЛ – T-клеточные неходжкинские лимфомы; ЛБ – лимфома Беркитта; ХЛЛ – хронический лимфоидный лейкоз; ХМЛ – хронический миелоидный лейкоз; ММ – множественная миелома; ЛХ – лимфома Ходжкина.
Note: PLCNS – primary lymphoma of the central nervous system, ALL – acute lymphoblastic leukemia, AML – acute myeloblastic leukemia, DL-BCL – diffuse large B-cell lymphoma, LMZS – lymphoma of the marginal zone of the spleen, B-NHL – B-cell non-Hodgkin lymphoma, T-NHL – T-cell non-Hodgkin lymphoma, BL – Burkitt lymphoma, CLL – chronic lymphocytic leukemia, CML – chronic myelogenous leukemia, MM – multiple myeloma, HL – Hodgkin’s lymphoma.
-
[45 ]. При этом было показано, что MIR-34A не метилирован в нормальных клетках крови, в отличие от клеточных линий ММ, лимфомы Ходжкина (ЛХ) и неходжкинских лимфом (НХЛ), что может свидетельствовать о специфичности гиперметилирования гена для лимфоидных клеток опухолевого происхождения и подтверждать онкосупрессорную
роль данной микроРНК [35, 46]. Согласно данным другого исследования, метилирование MIR-34B/C было выявлено в 34 % случаев ОЛЛ [47].
Таким образом, складывается впечатление, что гены кластера miR-34 часто гиперметилированы при лимфопролиферативных заболеваниях опухолеспецифическим образом.
Ген MIR-203
MIR-203 расположен в межгенном участке хромосомного локуса 14q32.33, который часто подвергается делеции при опухолях. Кодируемая им микроРНК miR-203 впервые была идентифицирована в коже, где она способствует дифференцировке клеток эпидермиса и ограничивает их пролиферативный потенциал [48]. В недавнем исследовании было показано, что преходящее воздействие этой микроРНК улучшает дифференци-ровочную способность мышиных и человеческих плюрипотентных стволовых клеток [49].
Было отмечено, что повышение экспрессии miR-203 происходит при активации р53. В основе этого явления лежит ускорение созревания miR-203 путем усиления ее Drosha-опосредованного процессинга под действием белка р53 [20]. Сообщалось также, что miR-203 нацелена на эволюционно консервативную последовательность в 3’-нетранслируемой области м-РНК BCL-W , который имеет антиапоптотическую активность. Так, усиленная экспрессия BCL-W делает лимфоидные и миелоидные клетки рефрактерными к цитотоксическим воздействиям. Однако индукция miR-203 приводит к подавлению экспрессии BCL-W и антиапоптотическому эффекту только в p53 (+) клетках [51].
Биоинформационный анализ показывает, что miR-203 может повышать радиационную чувствительность и миграционную способность клеток за счет посттранскрипционного контроля генов PI3K/AKT, SRC, Ras/MAPK и JAK/STAT3 сигнальных путей, а именно, за счет нацеливания на следующие мишени: ATM , RAD51 , SRC , PLD2 , FGF2 , PI3K-AKT , JAK-STAT3 , VEGF , HIF-1α и MMP2 . Также предсказанными мишенями miR-203 является ДНК-метилтрансфераза DNMT3b и мРНК гена CREB1 , который участвует в пролиферации и выживании лейкозных клеток.
MIR-203 имеет CpG островок протяженностью 804 п.н. [52]. Метилирование его как причина снижения экспрессии miR-203 показано при солидных опухолях [50, 53]. При лимфопролиферативных заболеваниях впервые метилирование гена MIR-203 показано при Т-клеточных лимфомах [54]. Частота данного события при ОЛЛ составляла 27 % [47].
Позднее была изучена экспрессия и метилирование MIR-203 в смешанной выборке онкогема- тологических больных (n=150). Метилирование обнаруживалось в образцах ОМЛ с частотой 10 %, хронического лимфоидного лейкоза (ХЛЛ) – 42 % и НХЛ – 38,8 % (в 60 % – при лимфомах из NK-клеток, в 40,9 % – при лимфомах В-клеточного, в 23,5 % – при Т-клеточного происхождения). В нормальных клетках крови и костного мозга метилирование MIR-203 выявлено не было, что согласуется с идеей об опухолеспецифичном паттерне метилирования гена данной микроРНК [55].
В опухолевых клетках больных хроническим миелоидным лейкозом (ХМЛ) и в линии бластных клеток Ph-позитивного ХМЛ K-562 метилирование промотора гена отсутствовало, что плохо согласовалось с данными предыдущих исследований о преимущественном гиперметилировании MIR-203 при Ph-позитивных лейкозах (с экспрессией BCR-ABL ) и редком его обнаружении при Ph-негативных острых лейкозах или хронических миелопролиферативных заболеваниях [54]. Также было показано, что ингибитор BCR-ABL иматиниб вызывает деметилирование промоторной области MIR-203 , что приводит к низкой экспрессии целевого гена BCR-ABL1 и остановке пролиферации лейкозных клеток [56].
Сопоставление частот фиксации аберрантного метилирования MIR-203 при моноклональных гаммапатиях неясного генеза, впервые диагностированной множественной миеломе и в рецидиве заболевания позволяет предположить, что гиперметилирование данного гена является ранним событием в генезе заболевания, а не приобретается на этапах прогрессирования опухоли [57].
На клеточных линиях клеток лимфомы Ходжкина, неходжкинских лимфом и множественной миеломы была показана обратимость аберрантного метилирования промотора MIR-203 под действием обработки 5-Аза-2'-дезоксицитидином, что приводило к восстановлению экспрессии miR-203. Нормализация уровня микроРНК в клетках ингибировала клеточную пролиферацию или индуцировала гибель опухолевых клеток, что подтверждало данные об ее онкосупрессорной роли при лимфоидных новообразованиях [46, 54–56].
Общие мишени р53-респонзивных онкосупрессорных микроРНК
Развитие кроветворения – это многоступенчатый четко регулируемый процесс, в ходе которого гемопоэтические клетки подвергаются пролиферации и линейной дифференцировке. Он требует включение сети транскрипционных факторов, которые на различных этапах активируются или ингибируются высокоорганизованным образом. За последние десятилетия ряд исследований показал, что микроРНК могут влиять на экспрессию регуляторных белков в клетке [1].
р53-респонзивные микроРНК связаны с контролем многочисленных внутриклеточных сиг-
Таблица 2/ table 2
Общие значимые для развития гемобластозов мишени р53-респонзивных онкосупрессорных микроРНК common significant for the development of hemoblastosis targets of p53-responsive oncosuppressive microRNas
Мишень/Target C-MYC CDK4/6 MDM2/4
BCL2/MCL1 CYCLIN-D1 DNMT3A/B
K-RAS
FOXP1
NOTCH1/2
VEGFA
STAT3
SOX2/4/9
miR-143 miR-145
miR-34а
miR-34в/с miR-203
Примечание: серым цветом отмечены молекулярные мишени микроРНК.
Note: The molecular targets of microRNAs are marked in gray.
нальных путей, определяющих самообновление клеток, пролиферацию, репарацию повреждений ДНК, программированную клеточную смерть, эпителиально-мезенхимальный переход и миграцию, противоопухолевый иммунный ответ и неоангиогенез. По данным литературы, частыми общими мишенями для микроРНК miR-34a, miR-34b/с, miR-145, miR-143 и miR-203 являются м-РНК ряда проонокогенов (табл. 2). Для того чтобы продемонстрировать глубину имеющихся между ними взаимосвязей, необходимо отдельно остановиться на некоторых из мишеней данных молекул.
Циклин-D1 – это белок, который у человека кодируется геном CCND1 . Совместно с Циклином-D2 данный белок образует комплекс с регуляторными субъединицами циклинкиназ CDK4 и CDK6. В свою очередь, комплекс циклинов с циклинки-назами активирует белок ретинобластомы Rb, что способствует переходу клеточного цикла из G1 в S-фазу. Избыточная экспрессия циклинов и циклинкиназ, ускоряющая прогрессию клеточного цикла и деление клеток, часто наблюдается в различных опухолях и может способствовать онкогенезу [58].
Транскрипционный фактор C-MYC прямо или косвенно активирует экспрессию генов клеточного цикла – CCND2 и циклинкиназ, и, таким образом, также обеспечивает переход клеточного цикла в контрольной точке G1/S фаз. Другим важным про-онкогенным свойством С-MYC является то, что с его участием в клетке останавливается апоптоз, вызванный белком p53.
BCL2 и MCL1 относятся к белкам семейства BCL2, соотношение которых в клетке регулирует проницаемость митохондриальной мембраны. Они играют важную роль в выживании клеток и негативной регуляции программированной клеточной смерти, что реализуется как за счет прямого ингибирования ими проапоптотических белков, так и за счет предотвращения активации каспазозависимого пути апоптоза, гиперэкспрессия данных молекул в лимфоцитах в сочетании с повышением уровня C-MYC доказанно приводит к развитию агрессивных лимфопролиферативных опухолей [59].
Другая общая мишень активируемых р53 микроРНК – MDM2, который является важным негативным регулятором белка р53. Белок MDM2 функционирует как убиквитинлигаза, способствующая деградации р53, и как ингибитор транскрипционной активации гена ТР53 [60].
Гены DNMT3А и DNMT3B кодируют ДНК-метилтрансферазы, которые участвуют в метилировании генома de novo, в т. ч. аберрантном метилировании оноксупрессорных генов и генов р53-респонзивных микроРНК. Недавние наблюдения показывают, что DNMT3B – это основной фермент, метилирующий области активных генов, а нарушение его работы является общим признаком таких заболеваний человека, связанных с хромосомной и геномной нестабильностью, как опухоли [61]. Соматические мутации DNMT3A часто встречаются при ОМЛ и других гематологических злокачественных новообразованиях.
Таким образом, представленный обзор литературы показывает, что частыми общими мишенями для активируемых р53 микроРНК являются м-РНК ряда проонкогенных молекул, а именно транскрипционных факторов, а также позитивных регуляторов клеточного цикла в контрольной точке перехода G1/S фаз, антиапоптотических белков, метилтрансфераз.
Многие из описанных микроРНК встроены в двойные отрицательные регуляторные петли или регуляторные петли с положительной обратной связью. В связи с чем следует отметить наличие положительных обратных связей между р53 и активируемыми им микроРНК, а также отрицательные обратные связи между р53-респонзивными микроРНК и C-MYC и ДНК-метилтрансферазами. Соответственно, даже небольшое изменение уровня экспрессии микроРНК может иметь больший фенотипический эффект за счет усиления регуляторного контура.
Заключение
Поскольку аберрантное метилирование ДНК при опухолях может приводить к транскрипционной репрессии онкосупрессорных молекул и зачастую носит ткане- и/или опухолеспецифичный характер, нами проведен анализ имеющихся в литературе данных о частоте и значении метилирования генов ряда р53-респонзивных онкосупрессорных микроРНК при опухолевых заболеваниях системы крови. Обзор уточняет современные представления о регуляторной сети белка р53 и активируемых им микроРНК, а также подчеркивает функциональную ассоциацию р53-респонзивных молекул.
Важно, что каждая м-РНК содержит несколько эволюционно консервативных сайтов связывания с различными микроРНК, а каждая микроРНК может нацеливаться на несколько генов в одном и том же пути и, таким образом, достигать еще более значимой модуляции своей активности. При этом miR-34a, miR-34b/с, miR-145, miR-143 и miR-203 имеют множество общих мишеней. Таким образом, аберрантное метилирование генов, кодирующих данные микроРНК, потенциально может дерегулировать несколько функционально связанных мишеней, вовлеченных в отдельный
Список литературы Метилирование генов Р53-респонзивных онкосупрессорных микроРНК при гемобластозах
- Bondada M.S., Yao Y., Nair V Multifunctional miR-155 Pathway in Avian Oncogenic Virus-Induced Neoplastic Diseases. Noncoding RNA. 2019; 5(1): 24. doi: 10.3390/ncrna5010024.
- JurjA., PopL., PetrushevB., Pasca S., Dima D., Frinc I., DeakD., Desmirean M., Trifa A., Fetica B., Gafencu G., Selicean S., Moisoiu V., Micu W.T., Berce C., SacuA., Moldovan A., Colita A., BumbeaH., TanaseA., Dascalescu A., ZdrengheaM., StiufiucR., LeopoldN., TeteanR., BurzoE., Tomuleasa C., Berindan-Neagoe I. Exosome-carried microRNA-based signature as a cellular trigger for the evolution of chronic lymphocytic leukemia into Richter syndrome. Crit Rev Clin Lab Sci. 2018; 55(7): 501-15. doi: 10.1080/10408363.2018.1499707.
- HannafonB.N., Ding W.Q. Functional Role of miRNAs in the Progression of Breast Ductal Carcinoma in Situ. Am J Pathol. 2019; 189(5): 966-74. doi: 10.1016/j.ajpath.2018.06.025.
- Holubekova V., Mendelova A., JasekK., Mersakova S., Zubor P., Lasabova Z. Epigenetic regulation by DNA methylation and miRNA molecules in cancer. Future Oncol. 2017; 13(25): 2217-22. doi: 10.2217/ fon-2017-0363.
- Klein U., LiaM., CrespoM., CrespoM., SiegelR., Shen Q., Mo T., Ambesi-ImpiombatoA., Califano A, MigliazzaA., BhagatG., Dalla-FaveraR. The DLEU2/miR-15a/16-1 cluster controls B cell proliferation and its deletion leads to chronic lymphocytic leukemia. Cancer Cell. 2010; 17(1): 28-40. doi: 10.1016/j.ccr.2009.11.019.
- Larrabeiti-EtxebarriaA., Lopez-SantillanM., Santos-Zorrozua B., Lopez-Lopez E., Garcia-Orad A. Systematic Review of the Potential of MicroRNAs in Diffuse Large B Cell Lymphoma. Cancers (Basel). 2019; 11(2): 144. doi: 10.3390/cancers11020144.
- CuiB., ChenL., Zhang S., MrazM., Fecteau J.F., Yu J., GhiaE.M., Zhang L., Bao L., Rassenti L.Z., Messer K., Calin G.A., Croce C.M., Kipps T.J. MicroRNA-155 influences B-cell receptor signaling and associates with aggressive disease in chronic lymphocytic leukemia. Blood. 2014; 124(4): 546-54. doi: 10.1182/blood-2014-03-559690.
- Baylin S.B., Jones P.A. Epigenetic Determinants of Cancer. Cold Spring Harb Perspect Biol. 2016; 8(9). doi: 10.1101/cshperspect. a019505.
- DaniunaiteK., DubikaityteM., GibasP., BakaviciusA., LazutkaR.J., UlysA., JankeviciusF., Jarmalaite S. Clinical significance of miRNA host gene promoter methylation in prostate cancer. Hum Mol Genet. 2017; 26(13): 2451-61. doi: 10.1093/hmg/ddx138.
- Strmsek Z., Kunej T. MicroRNA Silencing by DNA Methylation in Human Cancer: a Literature Analysis. Noncoding RNA. 2015; 1(1): 44-52. doi: 10.3390/ncrna1010044.
- Walter R.F., Vollbrecht C., Werner R., Wohlschlaeger J., Christoph D.C., Schmid K.W., Mairinger F.D. microRNAs are differentially regulated between MDM2-positive and negative malignant pleural mesothelioma. Oncotarget. 2016; 7(14): 18713-21. doi: 10.18632/on-cotarget.7666.
- WrightonK.H. Small RNAs: p53 makes microRNAs mature. Nat Rev Mol Cell Biol. 2009; 10(9): 580-1. doi: 10.1038/nrm2749.
- Voropaeva E.N., Pospelova T.I., Voevoda M.I., Maksimov V.N., Orlov Y.L., Seregina O.B. Clinical aspects of TP53 gene inactivation in diffuse large B-cell lymphoma. BMC Med Genomics. 2019; 12(2): 35. doi: 10.1186/s12920-019-0484-9.
- Аль-Ради Л.Г., БаряхЕ.А., Белоусова И.Э., Бессмельцев С.С., ВоробьевВ.И., Вотякова О.М., Губкин А.В., ДеминаЕ.А., ДоронинВ.А., Желудкова О.Г., Загоскина Т.П., Коробкин А.В., Кравченко С.К., КузьминА.А., Лопаткина Т.Н., ЛориеЮ.Ю., Луговская С.А., Менделеева Л.П.,МихайловаН.Б.,Моисеева Т.Н., Мухортова О.В., НикитинЕ.А., Османов Е.А., Пивник А.В., Поддубная И.В., Поспелова Т.И., Птуш-кин В.В., Самойлова О.С., Самочатова Е.В., Стадник Е.А., Стефанов Д.Н., Тумян Г.С., Шатохин Ю.В., Шмаков Р.Г. Клинические рекомендации по диагностике и лечению лимфопролиферативных заболеваний. М., 2014. С. 288. [Al'-Radi L.G., Baryakh E.A., Belouso-va I.E., Bessmel'tsev S.S., Vorob'ev V.I., Votyakova O.M., Gubkin A.V., De-minaE.A., Doronin V.A., Zheludkova O.G., Zagoskina T.P., KorobkinA. V., Kravchenko S.K., Kuz'min A.A., Lopatkina T.N., Lorie Yu.Yu., Lugov-skaya S.A., Mendeleeva L.P., Mikhailova N.B., Moiseeva T.N., Mukhor-tova O.V., Nikitin E.A., Osmanov E.A., Pivnik A.V., Poddubnaya I.V., Pospelova T.I., Ptushkin V.V., Samoilova O.S., Samochatova E.V., Stad-nik E.A., Stefanov D.N., Tumyan G.S., Shatokhin Yu.V., Shmakov R.G. Clinical guidelines for the diagnosis and treatment of lymphoproliferative diseases. Moscow, 2014. 288 p. (in Russian)].
- Lozano G. The oncogenic roles of p53 mutants in mouse models. Curr Opin Gen Dev. 2007; 17(1): 66-70. doi: 10.1016/j. gde.2006.12.003.
- Solé C., LarreaE., Di Pinto G., TellaetxeM., Lawrie C.H. miRNAs in B-cell lymphoma: Molecular mechanisms and biomarker potential. Cancer Lett. 2017; 405: 79-89. doi: 10.1016/j.canlet.2017.07.020.
- Solé C., Arnaiz E., Lawrie C.H. MicroRNAs as Bio-markers of B-cell Lymphoma. Biomark Insights. 2018; 13. doi: 10.1177/1177271918806840.
- SachdevaM., Zhu S., Wu F., Wu H., Walia V., Kumar S., Elble R., Watabe K., Mo Y.Y. p53 represses c-Myc through induction of the tumor suppressor miR-145. Proc Natl Acad Sci USA. 2009; 106(9): 3207-12. doi: 10.1073/pnas.0808042106.
- Pidíkova P., Reis R., Herichova I. miRNA Clusters with Down-Regulated Expression in Human Colorectal Cancer and Their Regulation. Int J Mol Sci. 2020; 21(13): 4633. doi: 10.3390/ijms21134633.
- Suzuki H.I., Yamagata K., Sugimoto K., Iwamoto T., Kato S., Miyazono K. Modulation of microRNA processing by p53. Nature. 2009; 460(7254): 529-33. doi: 10.1038/nature08199.
- Xu N., Papagiannakopoulos T., Pan G., Thomson J.A., KosikK.S. MicroRNA-145 regulates OCT4, SOX2, and KLF4 and represses pluri-potency in human embryonic stem cells. Cell. 2009; 137(4): 647-58. doi: 10.1016/j.cell.2009.02.038.
- Go H., Jang J.Y., Kim C.W., Huh J., Kim P.J., Jeon Y.K. Identification of microRNAs modulated by DNA hypomethylating drugs in extranodal NK/T-cell lymphoma. Leuk Lymphoma. 2020; 61(1): 66-74. doi: 10.1080/10428194.2019.1654096.
- Hao S., Huo S., Du Z., Yang Q., RenM., Liu S., Liu T., Zhang G. MicroRNA-related transcription factor regulatory networks in human colorectal cancer. Medicine (Baltimore). 2019; 98(15). doi: 10.1097/ MD.0000000000015158.
- Chen W.Y., Lang Z.Q., Ren C., Yang P., Zhang B. miR-143 acts as a novel Big mitogen-activated protein kinase 1 suppressor and may inhibit invasion of glioma. Oncol Rep. 2019; 42(3): 1194-204. doi: 10.3892/ or. 2019.7218.
- Manvati S., Mangalhara K.C., Kalaiarasan P., Chopra R., Agarwal G., Kumar R., Saini S.K., Kaushik M., Arora A., Kumari U., Bamezai R.N.K., Dhar P.K. miR-145 supports cancer cell survival and shows association with DDR genes, methylation pattern, and epithelial to mesenchymal transition. Cancer Cell Int. 2019; 19: 230. doi: 10.1186/ s12935-019-0933-8.
- Shen W.F., Hu Y.L., Uttarwar L., Passegue E., Largman C. Mi-croRNA-126 regulates HOXA9 by binding to the homeobox. Mol Cell Biol. 2008; 28(14): 4609-19. doi: 10.1128/MCB.01652-07.
- Tan L.P., Wang M., Robertus J.L., Schakel R.N., Gibcus J.H., DiepstraA., Harms G., Peh S.C., ReijmersR.M., Pals S.T., KroesenB.J., Kluin P.M., Poppema S., van den Berg A. miRNA profiling of B-cell subsets: specific miRNA profile for germinal center B cells with variation between centroblasts and centrocytes. Lab Invest. 2009; 89(6): 708-16. doi: 10.1038/labinvest.2009.26.
- Akao Y., Nakagawa Y., Kitade Y., Kinoshita T., Naoe T. Downregu-lation of microRNAs-143 and -145 in B-cell malignancies. Cancer Sci. 2007; 98(12): 1914-20. doi: 10.1111/j.1349-7006.2007.00618.x.
- Roehle A., Hoefig K.P., Repsilber D., Thorns C., Ziepert M., Wesche K.O., Thiere M., Loeffler M., Klapper W., Pfreundschuh M., Matolcsy A., Bernd H.W., Reiniger L., Merz H., Feller A.C. MicroRNA signatures characterize diffuse large B-cell lymphomas and follicular lymphomas. Br J Haematol. 2008; 142(5): 732-44. doi: 10.1111/j.1365-2141.2008.07237.x.
- FischerL., HummelM., KorfelA., LenzeD., JoehrensK., ThielE. Differential micro-RNA expression in primary CNS and nodal diffuse large B-cell lymphomas. Neuro Oncol. 2011; 13(10): 1090-8. doi: 10.1093/ neuonc/nor107.
- Xia H., Yamada S., Aoyama M., Sato F., Masaki A., Ge Y., Ri M., Ishida T., UedaR., UtsunomiyaA., Asai K., Inagaki H. Prognostic impact of microRNA-145 down-regulation in adult T-cell leukemia/lymphoma. Hum Pathol. 2014; 45(6): 1192-8. doi: 10.1016/j.humpath.2014.01.017.
- Wu H., Liu C., Yang Q., Xin C., Du J., Sun F., Zhou L. MIR145-3p promotes autophagy and enhances bortezomib sensitivity in multiple myeloma by targeting HDAC4. Autophagy. 2020; 16(4): 683-97. doi: 10.1080/15548627.2019.1635380.
- Liu J., Li M., Wang Y., Luo J. Curcumin sensitizes prostate cancer cells to radiation partly via epigenetic activation of miR-143 and miR-143 mediated autophagy inhibition. J Drug Target. 2017; 25(7): 645-52. doi: 10.1080/1061186X.2017.1315686.
- Liu S.Y., Li X.Y., Chen W.Q., Hu H., Luo B., Shi Y.X., Wu T.W., Li Y., Kong Q.Z., LuH.D., Lu Z.X. Demethylation of the MIR145 promoter suppresses migration and invasion in breast cancer. Oncotarget. 2017; 8(37): 61731-41. doi: 10.18632/oncotarget.18686.
- Chim C.S., Wong K.Y., Qi Y., Loong F., Lam W.L., Wong L.G., Jin D.Y., Costello J.F., Liang R. Epigenetic inactivation of the miR-34a in hematological malignancies. Carcinogenesis. 2010; 31(4): 745-50. doi: 10.1093/carcin/bgq033.
- Asmar F, Hother C., Kulosman G., Treppendahl M.B., Nielsen HM., Ralfkiaer U., Pedersen A., M0ller M.B., Ralfkiaer E., de Nully Brown P., Gr0nbœk K. Diffuse large B-cell lymphoma with combined TP53 mutation and MIR34A methylation: Another «double hit» lymphoma with very poor outcome? Oncotarget. 2014; 5(7): 1912-25. doi: 10.18632/ oncotarget.1877.
- Naghizadeh S., Mohammadi A., Duijf P.H.G., Baradaran B., Safarzadeh E., Cho W.C., Mansoori B. The role of miR-34 in cancer drug resistance. J Cell Physiol. 2020; 235(10): 6424-40. doi: 10.1002/ jcp.29640.
- Zhang L., Liao Y., Tang L. MicroRNA-34 family: a potential tumor suppressor and therapeutic candidate in cancer. J Exp Clin Cancer Res. 2019; 38(1): 53. doi: 10.1186/s13046-019-1059-5.
- Xiong S., HuM., Li C., ZhouX., ChenH. Role of miR-34 in gastric cancer: From bench to bedside (Review). Oncol Rep. 2019; 42(5): 1635-46. doi: 10.3892/or. 2019.7280.
- VanRoosbroeckK., Calin G.A. MicroRNAs in chronic lymphocytic leukemia: miRacle or miRage for prognosis and targeted therapies? Semin Oncol. 2016; 43(2): 209-14. doi: 10.1053/j.seminoncol.2016.02.015.
- Peng D., Wang H., Li L., Ma X., Chen Y., Zhou H., Luo Y., Xiao Y., LiuL. miR-34c-5p promotes eradication of acute myeloid leukemia stem cells by inducing senescence through selective RAB27B targeting to inhibit exosome shedding. Leukemia. 2018; 32(5): 1180-8. doi: 10.1038/ s41375-018-0015-2.
- Craig V.J., Cogliatti S.B., Imig J., Renner C., Neuenschwander S., Rehrauer H., Schlapbach R., Dirnhofer S., Tzankov A., Müller A. Myc-mediated repression of microRNA-34a promotes high-grade transformation of B-cell lymphoma by dysregulation of FoxP1. Blood. 2011; 117(23): 6227-36. doi: 10.1182/blood-2010-10-312231.
- Craig V.J., Tzankov A., Flori M., Schmid C.A., Bader A.G., Müller A. Systemic microRNA-34a delivery induces apoptosis and abrogates growth of diffuse large B-cell lymphoma in vivo. Leukemia. 2012; 26(11): 2421-4. doi: 10.1038/leu.2012.110.
- Leucci E., Cocco M., Onnis A., De Falco G., van CleefP., Bellan C., van Rijk A., Nyagol J., Byakika B., Lazzi S., Tosi P., van Krieken H., Le-onciniL. MYC translocation-negative classical Burkitt lymphoma cases: an alternative pathogenetic mechanism involving miRNA deregulation. J Pathol. 2008; 216(4): 440-50. doi: 10.1002/path.2410.
- ZaroneM.R., Misso G., Grimaldi A., Zappavigna S., RussoM., Am-lerE., DiMartinoM.T., AmodioN., TagliaferriP., TassoneP., CaragliaM. Evidence of novel miR-34a-based therapeutic approaches for multiple myeloma treatment. Sci Rep. 2017; 7(1): 17949. doi: 10.1038/s41598-017-18186-0.
- Navarro A., Díaz T., Cordeiro A., Beyá M.D., Ferrer G., Fuster D., Martinez A., Monzó M. Epigenetic regulation of microRNA expression in Hodgkin lymphoma. Leuk Lymphoma. 2015; 56(9): 2683-9. doi: 10.3109/10428194.2014.995650.
- Roman-Gomez J., Agirre X., Jiménez-Velasco A., Arqueros V., Vilas-Zornoza A., Rodriguez-Otero P., Martin-Subero I., Garate L., Cordeu L., San José-Eneriz E., Martin V., Castillejo J.A., Bandrés E., Calasanz M.J., Siebert R., Heiniger A., Torres A., Prosper F. Epigenetic regulation of microRNAs in acute lymphoblastic leukemia. J Clin Oncol. 2009; 27(8): 1316-22. doi: 10.1200/jœ.2008.19.3441.
- Chakraborty C., Sharma A.R., Patra B.C., Bhattacharya M., Sharma G., Lee S.S. MicroRNAs mediated regulation of MAPK signaling pathways in chronic myeloid leukemia. Oncotarget. 2016; 7(27): 42683-97. doi: 10.18632/oncotarget.7977.
- Salazar-RoaM., TrakalaM., Alvarez-FernándezM., Valdés-MoraF., Zhong C., Muñoz J., Yu Y., Peters T.J., Graña-Castro O., Serrano R., Zapatero-Solana E., Abad M., Bueno M.J., Gómez de Cedrón M., Fernández-Piqueras J., Serrano M., BlascoM.A., Wang D.Z., Clark S.J., Izpisua-Belmonte J.C., Ortega S., Malumbres M. Transient exposure to miR-203 enhances the differentiation capacity of established pluripotent stem cells. EMBO J. 2020; 39(16). doi: 10.15252/embj.2019104324.
- Braga E.A., Fridman M.V., Loginov V.I., Dmitriev A.A., Moro-zov S.G. Molecular Mechanisms in Clear Cell Renal Cell Carcinoma: Role of miRNAs and Hypermethylated miRNA Genes in Crucial Oncogenic Pathways and Processes. Front Genet. 2019; 10: 320. doi: 10.3389/ fgene.2019.00320.
- Funamizu N., Lacy C.R., Kamada M., Yanaga K., Manome Y. MicroRNA-203 induces apoptosis by upregulating Puma expression in colon and lung cancer cells. Int J Oncol. 2015; 47(5): 1981-8. doi: 10.3892/ijo.2015.3178.
- Schoof C.R.G., Izzotti A., Jasiulionis M.G., dos Reis VasquesL. The Roles of miR-26, miR-29, and miR-203 in the Silencing of the Epigenetic Machinery during Melanocyte Transformation. Biomed Res Int. 2015. doi: 10.1155/2015/634749.
- Benati M., Montagnana M., Danese E., Paviati E., Giudici S., Franchi M., Lippi G. Evaluation of mir-203 Expression Levels and DNA Promoter Methylation Status in Serum of Patients with Endometrial Cancer. Clin Lab. 2017; 63(10): 1675-81. doi: 10.7754/Clin.Lab.2017.170421.
- BuenoMJ., Pérez de Castro I., Gómez de CedrónM., Santos J., Calin G.A., Cigudosa J.C., Croce C.M., Fernández-Piqueras J., Malumbres M. Genetic and epigenetic silencing of microRNA-203 enhances ABL1 and BCR-ABL1 oncogene expression. Cancer Cell. 2008; 13(6): 496-506. doi: 10.1016/j.ccr.2008.04.018.
- Chim C.S., Wong K.Y., Leung C.Y., Chung L.P., Hui P.K., Chan S.Y., Yu L. Epigenetic inactivation of the hsa-miR-203 in haematological malignancies. J Cell Mol Med. 2011; 15(12): 2760-7. doi: 10.1111/j.1582-4934.2011.01274.x.
- Shibuta T., HondaE., Shiotsu H., Tanaka Y., Vellasamy S., Shirat-suchiM., Umemura T. Imatinib induces demethylation of miR-203 gene: an epigenetic mechanism of anti-tumor effect of imatinib. Leuk Res. 2013; 37(10): 1278-86. doi: 10.1016/j.leukres.2013.07.019.
- Wong K.Y., Liang R, So C.C., Jin D.Y., Costello J.F., Chim C.S. Epigenetic silencing of MIR203 in multiple myeloma. Br J Haematol. 2011; 154(5): 569-78. doi: 10.1111/j.1365-2141.2011.08782.x.
- Basso K., Saito M., Sumazin P., Margolin A.A., Wang K., Lim W.K., Kitagawa Y., Schneider C., Alvarez M.J., CalifanoA., Dalla-FaveraR. Integrated biochemical and computational approach identifies BCL6 direct target genes controlling multiple pathways in normal germinal center B cells. Blood. 2010; 115(5): 975-84. doi: 10.1182/blood-2009-06-227017.
- Zhao L., Samuels T., Winckler S., Korgaonkar C., Tompkins V., Horne M.C., Quelle D.E. Cyclin G1 has growth inhibitory activity linked to the ARF-Mdm2-p53 and pRb tumor suppressor pathways. Mol Cancer Res. 2003; 1(3): 195-206.
- GagliardiM., StrazzulloM., MatarazzoM.R. DNMT3B Functions: Novel Insights From Human Disease. Front Cell Dev Biol. 2018; 6: 140. doi: 10.3389/fcell.2018.00140.
- Veland N., Chen T. Mechanisms of DNA methylation and dem-ethylation during mammalian development. In Handbook of Epigenetics: The New Molecular and Medical Genetics. 2017; 1: 11-24. doi: 10.1016/ b978-0-12-805388-1.00002-x.
