МЕТОДИКА ОЦЕНКИ КАЧЕСТВА ГЕНОМНОЙ СБОРКИ НА ОСНОВЕ АНАЛИЗА ЧАСТОТНОСТИ K-МЕРОВ В СЕКВЕНАТОРЕ ПАРАЛЛЕЛЬНОГО СЕКВЕНИРОВАНИЯ

Автор: А. Г. Бородинов, В. В. Манойлов, И. В. Заруцкий, А. И. Петров, В. Е. Курочкин
Журнал: Научное приборостроение @nauchnoe-priborostroenie
Рубрика: Системный анализ приборов и измерительных методик
Статья в выпуске: 1 т.32, 2022 года.
Бесплатный доступ
В настоящее время в связи с развитием приборостроения для проведения генетического анализа существует острая необходимость в разработке методик оценки качества геномной сборки. Подсчет встречаемости различных k-меров часто возникает в задачах сборки генома. В данной работе на основе анализа различных программных средств выбраны программы, которые позволяют оценить качество геномной сборки. С помощью выбранных программ обработаны данные, полученные на отечественном секвенаторе параллельного секвенирования Нанофор СПС. На основе результатов обработки этих данных произведена оценка качества геномной сборки по методике анализа k-меров для прибора Нанофор СПС
K-мер, NGS-методы, биоинформаика, сборка генома
Короткий адрес: https://sciup.org/142231103
IDR: 142231103 | УДК: 543.51+ 681.2–5 | DOI: 10.18358/np-32-1-e107
METHODOLOGY FOR ASSESSING THE QUALITY OF GENOMIC ASSEMBLY BASED ON THE ANALYSIS OF THE FREQUENCY OF K-MERS IN A PARALLEL SEQUENCING SEQUENCER
Counting the occurrence of different k-mers often causes problems of genome assembly. Analysis of the frequency distribution of k-mers makes it possible to find assembly errors in already formed contigs. Currently, in connection with the development of instrumentation for genetic analysis, there is an urgent need to develop methods for assessing the quality of genomic assembly. Such techniques will make it possible to assess the reliability of genetic analysis in existing and newly developed devices. In this work, based on the analysis of various software tools, programs were selected to assess the quality of genomic assembly in parallel sequencing sequencers. Using the selected programs, the data obtained on the domestic sequencer for parallel sequencing Nanofor SPS were processed. Based on the results of processing these data, the quality of the genomic assembly was assessed by the method of analysis of k-mers and recommendations were given for improving the hardware and software of the Nanofor SPS device.
Текст научной статьи МЕТОДИКА ОЦЕНКИ КАЧЕСТВА ГЕНОМНОЙ СБОРКИ НА ОСНОВЕ АНАЛИЗА ЧАСТОТНОСТИ K-МЕРОВ В СЕКВЕНАТОРЕ ПАРАЛЛЕЛЬНОГО СЕКВЕНИРОВАНИЯ
K -мер — это просто последовательность из k символов в строке (или нуклеотидов в последовательности ДНК в задаче секвенирования). Разложение последовательности на ее k -меры позволяет анализировать этот набор фрагментов фиксированного размера, а не последовательность целиком, и это может быть более эффективным подходом. Простой пример: чтобы проверить, происходит ли последовательность S из организма A или из организма B, предполагая, что геномы A и B известны и достаточно разные, мы можем проверить, содержит ли S больше k -меров, присутствующих в A или в B.
Практически любой геном содержит повторяющиеся области, однако, начиная с определенного значения k , k -меры определенным образом однозначно идентифицируют его; если мы посчитаем количество появлений k -мер для достаточно большого k (ограниченного сверху длиной чтения), оказывается, что большинство из них находятся в геноме в единственном экземпляре. Например, если порядок длины генома сравним с человеческим, вероятность встретить случайную подстроку длины 14 хотя бы один раз составляет 0.975893 [1]. Для k = 20 эта же вероятность составляет 0.000909.
Подсчет встречаемости различных k-меров часто возникает в задачах сборки генома. Распределение частот встречаемости используется для процедуры корректирования ридов, что подразумевает разделение содержащихся k-меров на "доверенные" и "ошибочные" [1]. Подобная информация генома используется некоторыми программами сборки генома для определения того, является ли рассматриваемый участок повтором или нет.
В настоящее время в связи с развитием приборостроения для проведения генетического анализа существует острая необходимость в разработке методик оценки качества геномной сборки. Такие методики позволят оценить достоверность проведения генетического анализа в существующих и вновь разрабатываемых приборах. В данной работе на основе анализа различных программных средств выбраны программы, которые позволяют оценить качество геномной сборки в секвенаторах параллельного секвенирования. С помощью выбранных программ обработаны данные, полученные на отечественном секвенаторе параллельного секвенирования Нанофор СПС.
АНАЛИЗ ПРОГРАММНЫХ СРЕДСТВ ОЦЕНКИ КАЧЕСТВА СБОРКИ ГЕНОМА
Поскольку количество k -мер растет экспоненциально для значений k , подсчет k -мер для больших значений k является вычислительно сложной задачей. Хотя достаточно простые реализации работают для малых значений k , их необходимо адаптировать для приложений с высокой пропускной способностью или когда k велико. Для решения этой проблемы были разработаны различные инструменты:
-
• Jellyfish использует многопоточную хеш-таблицу без блокировок для подсчета k -мер и имеет реализации на Python, Ruby и Perl [2];
-
• KMC — это инструмент для подсчета k -мер, который использует многодисковую архитектуру для оптимизации скорости [3];
-
• Gerbil использует подход хеш-таблицы, но с дополнительной поддержкой ускорения графического процессора [4];
-
• K-mer Analysis Toolkit (KAT) использует модифицированную версию Jellyfish для анализа количества k -мер [5].
В качестве основного инструмента работы с k -мерами был выбран KAT (K-mer Analysis Tookit), представляющий эффективный набор средств для быстрого подсчета, сравнения и анализа спектров k -мер произвольной длины из данных генетических последовательностей.
Основным методом анализа при работе с k -мерами является проверка качества сборки генома путем сравнения характеристик k -меров совокупности анализируемых р и дов с референтным образцом или с собранным геномом (при сборке de novo ). Инструмент KAT hist — это графическое представление набора данных, показывающее, сколько коротких последовательностей фиксированной длины ( k -мер) появляется определенное количество раз. Частота встречаемости нанесена на ось х , а число k -меров на оси у . Пример 31-mer spectrum of S.cerevisae S288C WGS приведен на рис. 1.
Инструмент KAT comp генерирует матрицу с k-мерным набором последовательностей частот k-меров на одной оси, а частотой встречаемости k-меров другого набора на другой оси. При сравнении набора ридов со сборкой KAT сначала вычисляет свойства и состав k-меров сборки. При представлении в виде стоковых гистограмм спектр к-меров для ридов разбивается по числу копий к-меров для сборки. Кроме того, KAT предоставляет инструмент sect для отслеживания покрытия k-мерами, исходя из рассчитанных спектров k-меров для совокупности ридов и референса. Это может помочь идентифицировать такие артефакты сборки, как события сворачивания и разворачивания, или обнаруживать повторяющиеся области в последовательности ДНК.
KAT также включает инструмент hist для вычисления спектра из одного набора последовательностей и инструмент gcp для анализа гуанин-цитозин содержания (GC-контента) в зависимости от частоты k -меров. Инструмент filter можно использовать для выделения последовательностей из полного набора в соответствии либо с покрытием k -мерами или GC-содержанием для заданного набора. Эти инструменты могут использоваться для различных задач, включая обнаружение и извлечение загрязняющих веществ (contaminant detection) как в необработанных р и дах, так и в сборках (assemblies), анализ смещения по GC-составу и согласованность между парноконцевыми (paired end) р и дами с чувствительностью по концентрациям примесей от 0.1 ppm.
1 000 000
900 000 ■
800 000 ■
700 000 -. * •
600 000 ■ •'
Е• g 500 000
400 000 •
300 000 ■,
200 000-• юоооо ■ ,•
01-------------1-------------1-------------1-------------1f-
0 5 10 15 2025
30 35 40 45 50 55 60
Freq
Рис. 1. Графическое представление набора данных KAT hist
KAT прост в использовании, обеспечивает высокую скорость анализа. Время получения результатов анализа составляет не более минуты.
МЕТОДИКИ РАБОТЫ С K -МЕРАМИ
В работе [1] предложен метод оценки качества геномной сборки, заключающийся в установлении соответствия между уникальными k -мерами в собранном геноме и к -мерами в р и дах. Процедура выглядит следующим образом.
-
1. Построение гистограммы встречаемости k -меров для р и дов.
-
2. Выбор окрестности пика уникальных k -меров на гистограмме встречаемости.
-
3. Построение гистограммы встречаемости k -меров для каждой сборки.
-
4. Расчет меры Q как доли различных k -меров, взятых из окрестности пика на гистограмме встречаемости k -меров в чтениях.
-
5. Выбор сборки с максимальным значением Q в качестве наилучшей.
В работе [6] предложен метод исправления ошибок, оптимизированный для работы с чтениями, содержащими как ошибки замены, так и ошибки вставки и удаления. Поскольку ошибки происходят с небольшой частотой, вероятность того, что один и тот же k -мер будет прочитан несколько раз с одинаковым набором ошибок, очень мала. Из этого вытекает, что те k -меры, которые встречаются в наборе чтений мало раз, являются ошибочными, остальные же являются реальными подстроками генома (рис. 2).
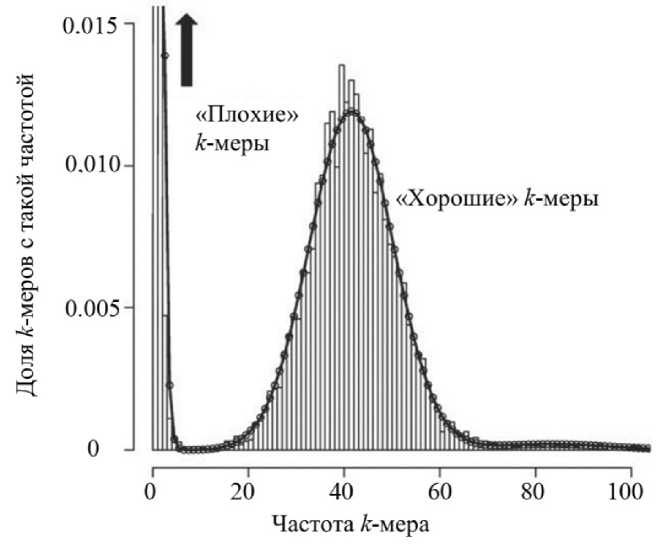
Рис. 2. Распределение частот к -меров в р и дах [6]
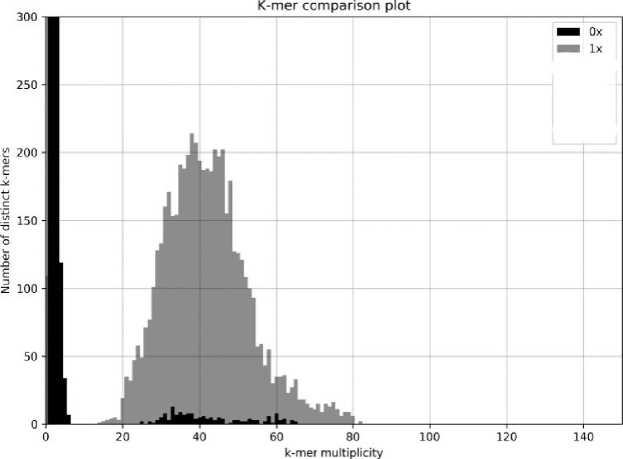
Рис. 3. Типичный k -mer comparison plot секвенирования Phix 174 на Illumina Miseq
ИСПОЛЬЗОВАНИЕ ПРОГРАММЫ KAT ДЛЯ ОБРАБОТКИ ДАННЫХ
СЕКВЕНАТОРА НАНОФОР СПС
Для обработки данных секвенатора Нанофор СПС была использована опция программы КАТ "K-mer comparison plot". По сути мы представляем, сколько элементов каждой частоты в спектре ридов оказались не включены в референтный ге- ном (в нашем случае Phix174), включены один раз, включены дважды и т.д.
На рис. 3, 4 представлены k-mer comparison plot, полученные соответственно для приборов Illumina и Нанофор СПС. Показательно, что для сходных характеристик проточных ячеек запуск Нанофор СПС обеспечивает больший уровень покрытия р и дами референсной последовательности (центр тяжести k -меров с уникальным покрытием).
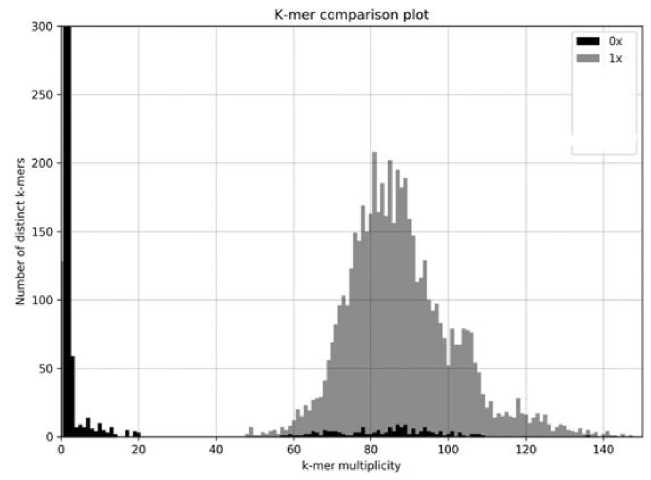
Рис. 4. Типичный k -mer comparison plot секвенирования Phix 174 на Нанофор СПС
ЗАКЛЮЧЕНИЕ
Проекты сборки генома обходятся дорого как по времени, так и по вложенным средствам. В этом случае выявление проблем с экспериментальными данными, обаруженных уже после сборки, может стать настоящей неудачей. С помощью K-mer Analysis Toolkit (KAT) исследователи могут получить доступ к качественным критериям и подтвердить свои результаты на более ранних этапах.
K -меры представляют собой небольшие фрагменты исходного генома с фиксированным числом оснований ДНК. Компьютер может эффективно работать с большим количеством k -меров, а затем идентифицировать связи между этими фрагментами, чтобы создать представление об исходном геноме. Основанные на k -мерах методы обычно используются для эффективного создания геномных сборок. KAT построен для изучения и сравнения наборов данных секвенирования с использованием основных свойств каждого отдельного k -мера, таких как частота встречаемости и нуклеотидный состав.
В первую очередь KAT может анализировать данные секвенирования для определения уровней случайных ошибок, систематических ошибок и контаминации. Информация, полученная в ходе этого анализа, может помочь исследователям решить, следует ли продолжать выполнение последующих задач, таких как сборка генома. Затем KAT может перепроверить проведенную сборку генома, определив полноту и точность сборки без каких-либо внешних справочных данных.
