Микрофлюидная жидкостная система экстракции с химически индуцированным образованием трех фаз в потоке

Автор: Сляднев М.Н., Казаков В.А., Макаров Е.Д., Ганеев А.А., Москвин Л. Н.
Журнал: Научное приборостроение @nauchnoe-priborostroenie
Рубрика: Материалы научного семинара "Микрочиповые технологии в аналитической химии"
Статья в выпуске: 2 т.15, 2005 года.
Бесплатный доступ
Предложена оригинальная система двух гомогенных растворов, представляющих собой смеси гептан-хлористый метилен-ацетонитрил и вода-ацетонитрил, при смешивании которых происходит образование трех сосуществующих фаз. Первая органическая фаза содержит преимущественно гептан, вторая (акцепторная) органическая фаза включает ацетонитрил, хлористый метилен и небольшое количество гептана, третья фаза состоит преимущественно из воды. При введении двух исходных фаз в микроканал чипа размерами 200 мкм (Ш) × 100 мкм (Г) показана возможность образования трехфазного потока со стабильной поверхностью раздела между всеми фазами. Проведено изучение процесса массообмена в образующейся трехфазной системе на примере распределения родамина 6G, включенного в состав начального водно-ацетонитрильного раствора, при введении в систему хлорида калия и без него. Для равновесных условий распределения родамина между фазами при смешивании начальных растворов в стеклянном цилиндре измерены аналитические характеристики процесса массообмена (коэффициенты концентрирования и распределения) при различных объемных соотношениях начальных растворов. Для динамических условий проведения экстракции родамина в микроканале чипа показана возможность протекания массообмена до равновесного состояния за время около 1 секунды с момента начала контакта начальных фаз. Малое время установления равновесия в микроканале чипа обусловлено диффузией вещества на малые расстояния в поперечном сечении микроканала, большим отношением площади межфазной границы к объему фаз, а также механизмом протекания массообмена в условиях выделения большей части ацетонитрила из исходного водно-органического раствора в отдельную фазу с одновременным переходом в нее молекул красителя.
Короткий адрес: https://sciup.org/14264377
IDR: 14264377 | УДК: 582.288:
Microfluidic liquid-liquid extraction system with a chemically induced three-phase flow
This paper deals with a liquid extraction system of two immiscible liquids, i.e. heptane-dichloromethane-acetonitrile and water-acetonitrile that after confluence form a three-phase system. Each of three chemically induced phases mainly consists of heptane (1st phase), acetonitrile/ dichloromethane (2nd phase) and water (3rd phase). stable interfaces between all of three phases were observed under flow conditions, when two initial phases were injected into a microchannel of 200(W)×100(D) μm. The mass transfer of the model compound (rhodamine 6G) between phases was studied in static conditions and analytical characteristics of the processes were obtained for different compositions of initial phases. For microchannel flow conditions, it was found that equilibrium was almost reached in a 1 second after the confluence of initial phases. This short time constant was attributed to the fast diffusion across the chemically induced phases in the microchannel, large area to volume ratio of the interfacial regions, and to the phase formation mechanism, which is characterized by the transfer of a large extent of acetonitrile from the initial phase to a chemically induced phase with simultaneous transfer of dye molecules.
Текст статьи Микрофлюидная жидкостная система экстракции с химически индуцированным образованием трех фаз в потоке
Одним из направлений применения микрофлю-идных микрочипов (МФЧ) в аналитической химии является осуществление процессов массообмена в гомогенных и мультифазных системах создания микрофлюидных потоков. Исследования диффузионного процесса массообмена в гомогенной системе были проведены на примере операций смешивания, реакции или диффузионной фильтрации веществ в микроканалах чипов [1–5]. Реализацией процессов массообмена в мультифазных системах является проведение жидкостной экстракции (ЖЭ) в системах двух несмешивающихся жидкостей [6– 11], твердофазной микроэкстракции [12–13], абсорбции и парофазного анализа в системах жидкость—газ [14–17], а также диализа веществ через жидкую [18] или синтезированную органическую мембрану [19]. Проведение подобных процессов и операций, как правило, востребовано при реализации стадии пробоподготовки/дериватизации в МФЧ при создании интегрированных лабораторий на чипе для анализа веществ.
Как было показано в работах [10, 11, 18, 20–22], при осуществлении операций массообмена в МФЧ за счет ламинарного режима микрофлюидных по- токов достигается образование стабильного интерфейса между сосуществующими фазами и наблюдается преимущественно диффузионный характер массообмена. Математический аппарат для теоретического описания протекающих процессов основан на использовании законов диффузии Фика [23]. При этом полнота протекания процессов массообмена до равновесного состояния определяется временем контакта микрофлюидных потоков в микроканалах чипов.
При осуществлении операций массообмена в жидкостных системах время, необходимое для равновесного протекания процессов в микроканалах чипов с типичными геометрическими размерами порядка десятков и сотен микрометров, составляет десятки и сотни секунд и зависит от скорости диффузии веществ и природы применяемых растворителей [6–11, 18]. Хотя по сравнению с обычными макросистемами с линейными размерами порядка единиц и десятков сантиметров время достижения равновесия в условиях диффузионного распределения веществ в МФЧ чрезвычайно мало, в этих условиях для практического достижения подобных времен контакта необходимо создавать микрофлюидные потоки со скоростью порядка десятков и сотен пл/с, а также увеличи- вать длину микроканалов до нескольких десятков сантиметров [6 –11]. При этом возникают трудности, связанные с устойчивостью межфазной границы для мультифазных систем, а также с технической реализацией создания микрофлюидных потоков.
Ускорения протекания процессов массообмена в мультифазных жидкостных системах, по-видимому, можно достичь при создании системы микрофлюидных потоков, в которой происходит химически-индуцированное образование новой акцепторной фазы в микроканале чипа. При этом в зависимости от состава первоначальных жидкостных потоков и соотношения их объемных скоростей возникает возможность контроля ширины зоны этой фазы/(объемной скорости) в микроканале МФЧ и, таким образом, контроля полноты протекания диффузионного массообмена в поперечном сечении микроканала. Нами предложена система двух жидкостей, при контакте которых происходит образование подобной трехфазной системы экстракции (ТФСЭ).
Настоящая статья посвящена изучению условий образования ТФСЭ при смешивании двух гомогенных растворов: вода—ацетонитрил и гептанхлористый метилен—ацетонитрил в стационарных и динамических условиях. В качестве модельного вещества для изучения кинетики фазообразования и полноты протекания массообмена использован краситель родамин 6G, введенный в состав ацетонитрильно-водной фазы. Проведено изучение условий образования ТФСЭ в стационарных условиях при смешивании начальных фаз определенного состава в различном объемном соотношении в стеклянном цилиндре и определены аналитические характеристики массообмена модельного вещества (коэффициенты концентрирования и распределения) в этих условиях. Показана возможность проведения экстракции родамина в образующуюся акцепторную фазу в динамических условиях в микроканале МФЧ за короткий промежуток времени.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Растворы, реактивы и оборудование
Исходный 10–2 М концентрированный раствор родамина 6G готовили растворением точной навески соответствующего вещества в 95 %-м этаноле. Раствор хранили в холодильнике при + 4 ºС и использовали в течение месяца. Рабочие водноорганические растворы красителя готовили перед измерениями разбавлением исходного ацетонитрилом и водой с введением в растворы солевой добавки хлорида калия или без солевой добавки. Концентрация красителя в рабочих растворах составляла 5.72∙10–4 М при концентрации солевой добавки (KCl) 300 мМ. Объемная доля ацетонитрила в растворе при этом составляла 45 %.
Для оценки аналитических характеристик установки флуоресцентного детектирования использовали свежеприготовленные спиртовые растворы родамина 6G в диапазоне концентраций 5∙10–7÷ 2∙10–3 М.
Органическая фаза для создания ТФСЭ представляла собой свежеприготовленную смесь органических растворителей (гептан—хлористый ме-тилен—ацетонитрил в объемном соотношении 10:2.5:1). В работе использовали органические растворители квалификации не ниже "х.ч." (Нева-Реактив).
Перед проведением экспериментов в МФЧ или в стационарных условиях водно-ацетонит-рильный раствор родамина и органическую смесь растворителей в пластиковых пробирках объемом 1.5 мл размешивали на центрифуге "5415D" (Eppendorf) при скорости 10 000 об/мин в течение 2 мин и дегазировали под вакуумом, создаваемым водоструйным насосом, в течение 3 мин.
Для определения состава образующихся фаз при смешивании трехкомпонентных органических смесей растворителей с водно-органическими растворами использовали газовый хроматограф "Цвет-100" с термостатируемой ( Т =60 ºС) капиллярной кварцевой колонкой (30 м × 0.32 мм) и плазменно-ионизационным детектором (ПИД). На стенки хроматографической колонки была нанесена неполярная полидиметилсилоксановая неподвижная фаза марки "Chrompack CP Sil-13 CB" (Chrompack). В качестве газа-носителя использовали азот, расход газа-носителя при проведении измерений составлял 1 мл/мин. Дозирование проб осуществляли шприцем МШ-1; объем пробы составлял 0.2 мкл.
Рис. 1. Топология и габаритные размеры стеклян-но-кремниевого Н-образного микрочипа
Экспериментальная установка
Топология использованного в работе Н-образного стеклянно-кремниевого МФЧ и его габаритные размеры показаны на рис. 1, а технология его изготовления подробно описана в работе [11]. Размеры микроканалов трапециевидного поперечного сечения составляли 250 мкм (верхнее основание) × 90 мкм (нижнее основание) ×110 мкм (высота трапеции). Длина центрального микроканала контакта двух микрофлюидных потоков жидкостей составляла 45 мм.
Экспериментальная микрочиповая установка состояла из трех блоков: системы флуоресцентного детектирования, системы создания и контроля микрофлюидных потоков жидкостей в МФЧ и подвижной двухкоординатной платформы для держателя микрочипа.
Система флуоресцентного детектирования аналогична описанной в статье [11], за исключением используемой камеры и спектральных характеристик элементов оптической схемы. В качестве детектора флуоресцентного излучения выбрана камера (Electron-Optronic) на основе термоэлектрически охлаждаемой ПЗС-матрицы (до -10 ° С). Камера обеспечивала цифровую передачу изображения в ПЭВМ с разрядностью 12 бит при разрешении изображения 752 × 582 пикселей. Управление режимами камеры (температурой, экспозицией 0.16–5 с, усилением сигнала) осуществлялось через последовательный порт.
Источником возбуждения флуоресценции выбран светодиод (Lumileds Lighting), излучающий в диапазоне 490-600 нм, A max = 53 0 нм, с мощностью светового потока 120 мВт. Для спектральной фильтрации излучения использовались: интерференционный светофильтр с полосой пропускания 505–550 нм, дихроичное зеркало с полосой пропускания 560–800 нм и интерференционный светофильтр с полосой пропускания 570–640 нм (Omega Optical).
Интегральное увеличение оптической системы установки детектирования составляло 2.4 (поле наблюдения 1.6 × 1.2 мм), а разрешающая способность — величину ~2.1 мкм/пиксел.
Система создания микрофлюидных потоков жидкостей представляла собой блок, состоящий из двух шприцевых насосов (Люмэкс), описанный в работе [11]. Насосы использовались в нагнетательном режиме работы. Рабочий диапазон объемных скоростей микрофлюидных потоков, создаваемых шприцевыми насосами, составлял 0.2– 20.0 мкл/с.
Держатель с микрочипом в сборе был закреплен на подвижной двухкоординатной платформе. Платформа использовалась для точной юстировки положения изучаемой зоны канала относительно фокуса оптической системы. Держатель также служил для подвода жидкостных потоков к микрочипу и их отвода во внешние сливные емкости [11].
РЕЗУЛЬТАТЫ И ОБСУЖДЕНИЕ
Изучение условий образования трехфазной системы и распределения красителя в ней в стационарных условиях
Образование трехфазной системы было замечено нами при контакте гомогенного раствора состава гептан—хлористый метилен—ацетонитрил (Ф1) с водно-ацетонитрильным раствором (Ф2), как показано на (рис. 2, а). В качестве модельного вещества для изучения процессов массообмена в такой системе, исходя из спектральных характеристик установки флуоресцентного детектирования, был выбран краситель родамин 6G, вводимый в состав фазы Ф2. Родамин 6G из-за своих
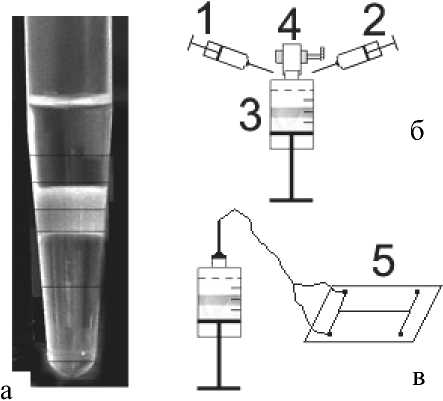
Рис. 2. Трехфазная система экстракции (ТФСЭ) в стационарных условиях.
а — трехфазная жидкостная система (модельное вещество: родамин 6G, добавка KCl в вод-но-ацетонитрильную фазу). Сверху вниз: неполярная органическая фаза, акцепторная органическая фаза, обедненная красителем водная фаза.
б — образование ТФСЭ; 1, 2 — шприцы для ввода начальных фаз; 3 — стеклянный шприц для определения объемов образующихся фаз ( 0 15 мм); 4 — силиконовый шланг и винтовой зажим для предотвращения испарения органических растворителей при установлении равновесия в системе.
в — поочередный ввод образовавшихся равновесных фаз в микроканалы чипа (5) для измерения аналитического сигнала химических свойств преимущественно переходит из первоначального ацетонитрильно-водного раствора в среднюю образующуюся фазу (см. рис. 2, а) и служит в качестве флуоресцентного маркера этой фазы. Изучение условий образования такой системы в стационарных условиях проводили с помощью установки, показанной на рис. 2, б. После ввода первоначальных фаз в стеклянный шприц для предотвращения испарения с помощью винтовых прижимов и силиконовых шлангов проводили герметизацию образующейся трехфазной системы, как показано на рис. 2, б, и считали протекающие процессы распределения родамина в ТФСЭ равновесными при времени контакта фаз с момента их слияния 1 час. При этом объемы первоначальных фаз составляли 4.4 (8.8) мл для Ф1 и 4.4 мл для Ф2, а площадь контакта фаз была равна 176.6 мм2.
При этом, судя по распределению красителя в системе и плотности используемых растворителей, верхняя органическая фаза преимущественно содержала гептан и, следовательно, была неполярной; средняя (акцепторная по отношению к красителю) органическая фаза включала растворители средней и высокой полярности: ацетонитрил и хлористый метилен; нижняя фаза состояла преимущественно из воды (рис. 2, а). В табл. 1 приведены объемы образующихся фаз при объемных соотношениях начальных фаз 1:1 и 2:1 (Ф1:Ф2). Для более четкого определения границ раздела трех фаз производили повышение коэффициента распределения родамина в образующуюся акцеп- торную фазу (АФ) за счет эффекта высаливания. С этой целью в первоначальный ацетонитрильноводный раствор родамина производили добавку хлорида калия. При СKCl =300 мМ при прочих равных условиях наблюдали повышение объема образующейся АФ за счет увеличения объемной доли ацетонитрила в ней (табл. 1). При этом, как видно из таблицы, при объемном соотношении начальных фаз 2:1 (Ф1:Ф2) происходило значительное уменьшение объема образующейся АФ по отношению к условиям равных объемов фаз с одновременным увеличением объема верхней органической фазы с большим содержанием гептана (рис. 2, а). Эффект солевой добавки, приводящий к увеличению объема АФ при равных объемах начальных фаз на ~9 %, приводил к увеличению объема АФ на 35 % при условии двукратного увеличения объема исходной органической фазы. При введении солевой добавки в систему наблюдали также значительно более быструю кинетику фазо-образования и одновременного распределения родамина в ТФСЭ.
С точки зрения возможности применения ТФСЭ в аналитической практике ключевую роль играет состав образующейся АФ. В табл. 1 приведен состав АФ при проведении экстракции родамина в статических условиях для объемного соотношения начальных фаз 1:1 и 2:1 (Ф1:Ф2). Состав АФ был определен с помощью газохроматографического анализа методом внутренней нормализации. При этом считали, что количество воды, которое невозможно измерить с помощью ПИД
Табл. 1. Объемы образующихся фаз и состав акцепторной фазы при различных объемных соотношениях начальных фаз в условиях солевой добавки в систему и без нее
|
V Ф1, мл |
V Ф2, мл |
С KCl в Ф2, мМ |
V АФ, мл |
V ОВФ, мл |
V ВОФ, мл |
Состав АФ, объем. % |
|
4.4 |
4.4 |
0 |
2.51 |
2.32 |
3.97 |
К1 — 10.8 ± 1, К2 — 36.6 ± 3, К3 — 52.6 ± 1 |
|
300 |
2.74 |
2.30 |
3.76 |
— |
||
|
8.8 |
4.4 |
0 |
1.50 |
2.30 |
9.40 |
К1 — 22.5 ± 2, К2 — 20.2 ± 3, К3 — 57.3 ± 2 |
|
300 |
2.03 |
2.13 |
9.04 |
— |
Примечание. К1 — гептан, К2 — хлористый метилен, К3 — ацетонитрил.
Ф1 — начальная органическая фаза (К1:K2:K3 = 10:2.5:1 ( V / V )); Ф2 — начальная водно-органическая фаза (К3:K4 = 1:1.25 ( V / V , К4 — вода)); АФ — акцепторная фаза, ОВФ — обедненная красителем водная фаза, ВОФ — верхняя органическая фаза (рис. 2, а)
газового хроматографа, в образующейся АФ пренебрежимо мало, что косвенно может быть подтверждено высоким содержанием гептана и хлористого метилена в этой фазе. Тем не менее, как видно из табл. 1, в составе АФ превалируют растворители высокой и средней полярности (ацетонитрил, хлористый метилен), объемная доля которых составляет более 80 %, что определяет полярность всей АФ. Ацетонитрил и хлористый метилен являются широко распространенными растворителями в аналитической практике, в том числе они находят широкое применение при анализе флуоресцирующих форм аналитов, квантовый выход которых в такой среде может быть в несколько раз выше по сравнению с водной [24, 25]. При этом полярность фазы, в приближении принимаемая равной полярности чистого ацетонитрила, способствует преимущественному переходу в нее широкого круга анализируемых соединений.
Поведение четырехкомпонентной системы состава "гептан—хлористый метилен—ацетонит-рил—вода" в зависимости от составов исходных фаз без учета роли солевой добавки можно прогнозировать с помощью построения трехмерной диаграммы расслаивания [26, 27]. Она представляет собой тетраэдр, вершины которого составляют чистые компоненты, по ребрам отложены составы бинарных систем, а грани являются диаграммами расслаивания трехкомпонентных смесей (треугольники Гиббса—Розебома [28]). С помощью построения такой диаграммы можно определить область сосуществования трех фаз. При этом объемы и составы образующихся фаз строго заданы составами исходных растворов и их объемным соотношением. Построение подобной диаграммы на основании данных газохроматографического анализа является предметом отдельного физикохимического исследования.
Аналитический сигнал (АС) от образующихся фаз измеряли с помощью установки флуоресцентного детектирования в микроканале МФЧ. Для этого осуществляли поочередный ввод первоначальных и образующихся фаз в микрочип с помощью шприца, как показано на (рис. 2, в). Время экспозиции кадра изображения при проведении измерений составляло 0.48 с. Такое время экспозиции было обусловлено перспективой изучения процессов массопереноса в МФЧ в быстро меняющихся динамических условиях проведения экстракции. Перед проведением измерений система детектирования при заданном времени экспозиции была откалибрована путем ввода спиртовых растворов с известной концентрацией красителя в микроканалы чипа. Целью калибровки являлся выбор рабочей концентрации родамина в начальном ацетонитрильно-водном растворе с точки зрения аналитических характеристик детектора. При этом для texp = 0.48 с линейный дипазон опреде- ляемых концентраций по родамину составлял 2∙10–5 ÷ 8∙10–4 М. На основании этих данных рабочая концентрация родамина составляла 5.72× ×10–4М.
Квантовый выход флуоресценции родамина ( φ R6G ) в спиртовом растворе на основании литературных данных [29, 30] составляет 0.95. При переходе от спирта к более полярным растворителям (ацетонитрилу и водным растворам) наблюдается изменение φ R6G и "синий" сдвиг спектров флуоресценции родамина 6G, что объясняется химическим строением молекулы красителя [31–33]. Поэтому при проведении измерений АС от фаз ТФСЭ учитывали влияние среды на флуоресценцию молекул красителя. Для этой цели в предварительных экспериментах проводили измерение АС в микроканале чипа от растворов с одинаковой концентрацией красителя в чистом ацетонитриле, 14 мМ боратном буферном растворе (рН 8.0) и спирте. Условные величины интенсивности флуоресценции (Fluorescence Intensity, FI) для ацетонитрильного раствора родамина и водного буферного раствора красителя составили соответственно 0.90 и 0.82 по отношению к FI красителя в спирте.
Коэффициенты концентрирования родамина после образования ТФСЭ рассчитывали по уравнению (1) и принимали равными отношению АС от образующейся АФ к АС от исходной донорной фазы (ИДФ) (Ф2) в микроканале чипа с учетом объемного соотношения этих фаз (при этом FI родамина в этих фазах принимали равной этой величине в чистом ацетонитриле):
K кS
(АС АФ / V АФ ) , (АС ИДФ / V ИДФ )
где АС АФ , АС ИДФ — интенсивности флуоресценции соответственно в акцепторной фазе и исходной донорной фазе, измеренные в микроканале чипа (усл. ед.); V АФ , V ИДФ — объемы акцепторной и исходной донорной фаз (мл).
Значения коэффициентов концентрирования для объемных соотношений исходных растворов 1:1 и 2:1 (Ф1:Ф2) приведены в табл. 2. Как видно из таблицы, коэффициенты концентрирования слабо зависят от введения солевой добавки в раствор ИДФ и возрастают с увеличением объемной доли Ф1 в начальной системе фаз. Это объясняется увеличением объема АФ при введении солевой добавки в систему и значительным уменьшением объема АФ при увеличении доли Ф1 в начальной системе (табл. 1). Полученные значения коэффициентов концентрирования являются подтверждением возможности проведения экстракции с одновременным концентрированием аналитов в АФ. Для повышения коэффициентов концентрирования (распределения) веществ в какую-либо из двух образующихся органических фаз (рис. 2, а),
Табл. 2. Значения коэффициентов концентрирования родамина R6G в образующуюся АФ и коэффициентов распределения красителя между АФ и ОВФ при различных объемных соотношениях начальных фаз в условиях солевой добавки в систему и без нее
|
V Ф1, мл |
V Ф2, мл |
С KCl в Ф2, мМ |
* K K S |
** K P S |
|
4.4 |
4.4 |
0 |
1.98 |
2.57 |
|
300 |
1.98 |
13.48 |
||
|
8.8 |
4.4 |
0 |
3.24 |
2.57 |
|
300 |
3.10 |
43.06 |
Примечание. В обозначениях К1 — гептан, К2 — хлористый метилен, К3 — ацетонитрил, К4 — вода:
Ф1 — начальная органическая фаза (К1:K2:K3 = 10:2.5:1 ( V / V ));
Ф2 — начальная водно-органическая фаза (К3:K4 = 1:1.25 ( V / V ));
C R6G в Ф2 = 5.72∙10–4 М.
*— расчет K KS по формуле (1); **— расчет K PS по формуле (2).
помимо оптимизации составов исходных фаз и их объемного соотношения, на наш взгляд, можно провести подбор условий протекания экстракции, например по ион-парному механизму, что является предметом отдельного исследования.
Коэффициенты распределения ( K PS ) родамина в ТФСЭ между двумя нижними фазами (рис. 2, а) рассчитывали по уравнению (2):
S (АС АФ / V АФ )
Kp — n р X
(АС ОВФ / V ОВФ)
где АС АФ , АС ОВФ — интенсивности флуоресценции в акцепторной фазе и обедненной красителем водной фазе, измеренные в микроканале чипа (усл. ед.); V АФ , V ОВФ — объемы акцепторной и исходной донорной фаз (мл); n р = 0.91.
KP S принимали равными отношению АС от образующейся АФ к АС от обедненной красителем водной фазы (ОВФ) в микроканале чипа с учетом объемного соотношения этих фаз и величин FI в них. При этом FI красителя для АФ принимали равной этой величине в чистом ацетонитриле, а FI родамина для ОВФ принимали равной этой величине в водном буферном растворе. Величина n P = FI ОВФ /FI АФ в уравнении (2) выражает отношение интенсивности флуоресценции родамина в этих растворах при равной концентрации красителя.
Значения KPS для объемных соотношений исходных растворов 1:1 и 2:1 (Ф1:Ф2) приведены в табл. 2. Как видно из таблицы, KPS родамина между АФ и ОВФ в образующейся ТФСЭ без соле- вой добавки не зависели от объемного соотношения исходных фаз, что можно объяснить постоянством составов АФ и ОВФ, поскольку коэффициент распределения вещества не зависит от объема фаз. При этом при увеличении соотношения Ф1:Ф2, как видно из табл. 1, происходило образование АФ примерно одинакового состава по основному компоненту (ацетонитрилу), а объем ОВФ оставался неизменным. Это говорит о постоянстве состава этой бинарной системы (ОВФ), поскольку образование ТФСЭ связано с перераспределением ацетонитрила в начальной системе фаз. Добавка KCl в ИДФ (Ф2) способствовала значительному повышению KPS. Это связано с эффектом высаливания органических компонентов из образующейся нижней водной фазы с высоким солевым фоном (рис. 2, а).
Следует отметить, что при проведении измерений АС от верхней неполярной фазы (рис. 2, а) оставался низким, что было связано с потерей флуоресцирующих свойств молекул родамина в среде с большой объемной долей гептана и большим объемом этой фазы (табл. 1).
Таким образом, путем варьирования составов контактирующих начальных фаз и их соотношения возникает возможность изменения объема образующейся АФ, ее полярности и других физических свойств, а также аналитических характеристик массообмена веществ (коэффициентов концентрирования и распределения) в образующейся ТФСЭ. Это может приводить к появлению новых нестандартных решений задач пробоподготовки и определения аналитов. Привлекательным в этом аспекте является проведение подобных исследований с помощью МФЧ.
Отметим, что с точки зрения целесообразности проведения пробоподготовки путем разбавления исходной пробы ацетонитрилом (или другим растворителем) с последующим выделением большей части этого растворителя и анализируемого вещества из материнского раствора в отдельную фазу эффект концентрирования аналита в образующуюся АФ по отношению к его концетрации в исходной неразбавленной пробе может быть невелик. Однако повышения чувствительности определения аналита можно достичь не только проведением операций по повышению уровня его концентрации, но и изменением характеристик анализируемого вещества, например значительным повышением квантового выхода флуоресцирующих форм в органической среде [24]. При этом следует отметить, что люминесцентные методы анализа из-за высокой чувствительности и технических характеристик систем детектирования нашли наиболее широкое применение в области микрофлю-идных аналитических систем по сравнению с другими [34].
Изучение условий образования трехфазной системы и распределения красителя в ней в потоке в микрофлюидном чипе
При проведении экстракции в динамических условиях в МФЧ для осуществления и стабилизации непрерывного "струйного" течения образующихся трех фаз в микроканале чипа (рис. 3, а) и предотвращения образования разрывов фазы АФ ("пробок") варьировали скорости микрофлюидных потоков двух начальных фаз и их соотношение.
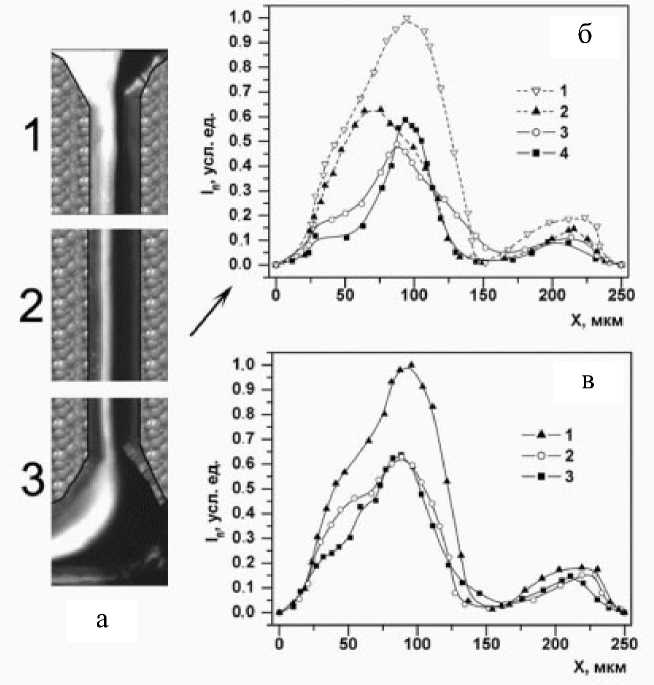
Рис. 3. Трехфазная система экстракции (ТФСЭ) в динамических условиях. а — образование ТФСЭ и кинетика распределения родамина 6G в системе по длине микроканала чипа при С KCl = 300 мМ (1, 2, 3).
б —профили интенсивности флуоресценции в поперечном сечении микроканала чипа при С KCl = 300 мМ в зависимости от времени контакта начальных фаз ( T конт ): 1 — 0 с; 2 — 0.1 с; 3 — 0.25 с; 4 — 1.12 с.
в — профили интенсивности флуоресценции в поперечном сечении микроканала чипа без добавки KCl в зависимости от времени контакта начальных фаз ( T конт): 1 — 0 с; 2 — 0.05 с; 3 — 1.12 с
При варьировании соотношения объемных скоростей подачи растворов Ф1 и Ф2 в МФЧ от 1:1 до 2:1 из-за физических свойств используемых жидкостей, топологии, размеров и формы поперечного сечения микроканалов чипа ширина зоны ИДФ (Ф2) в точке слияния начальных фаз в центральном микроканале чипа (рис. 1) изменялась от 60.0±0.5 до 57.0±0.5 % от максимальной ширины микроканала чипа. Измерения ширины зоны фаз Ф1 и Ф2 проводили после получения экспериментальных кадров оцифрованного изображения микроканала и построения профилей АС в его поперечном сечении [11]. Время экспозиции кадра изображения составляло 0.48 с. Оптимальные для стабилизации непрерывного течения трех образующихся фаз в микроканале МФЧ скорости мик-рофлюидных потоков составили 55 мкл/мин для органической фазы и 27 мкл/мин для ацетонитрильно-водного раствора родамина (соотношение скоростей подачи растворов в МФЧ 2:1). Поэтому для интерпретации полученных результатов по распределению родамина в динамических условиях с точки зрения равновесности протекания процессов использовали результаты по изучению распределения красителя в ТФСЭ в стационарных условиях при соотношении объемов начальных фаз 2:1 (Ф1:Ф2). При этом с помощью установки флуоресцентного детектирования наблюдали кинетику образования ТФСЭ и перехода родамина в АФ по длине микроканала чипа при добавке KCl в ИДФ или без нее.
На рис. 3, а показаны полученные экспериментальные кадры протекающих процессов массооб-мена по длине микроканала чипа при СKCl = 300 мМ в ИДФ. На рис. 3, б приведены профили аналитического сигнала в поперечном сечении микроканала для этих условий. Профили АС были получены путем математической обработки экспериментальных оцифрованных кадров изображения. Как видно из рис. 3, а (1, 2, 3), образование АФ происходило близко к середине микроканала чипа. По длине микроканала в этом месте постепенно формировалась зона шириной порядка 65 мкм с высоким значением АС, а также слева от нее зона обедненной красителем водной фазы (ОВФ) и справа зона неполярной органической фазы. Рост АС по краям микроканала в его поперечном сечении (рис. 3, б) был обусловлен эффектом отражения флуоресцентного излучения от образующейся АФ от зеркальных наклонных поверхностей кремниевых стенок [11]. По-видимому, ширина зоны АФ определена физическими свойствами жидкостей, а также размерами и формой микроканала. При проведении серии экспериментов с изменением объемной доли ацетонитрила в ИДФ от 20 до 50 % было замечено, что ширина зоны АФ в отличие от кинетики фазообра- зования при этом менялась слабо.
На рис. 3, в приведены профили АС в поперечном сечении микроканала МФЧ для условий образования ТФСЭ без добавки KCl. При этом, как видно из рис. 3, в, из-за низких коэффициентов распределения родамина в образующуюся АФ невозможно четко определить границу раздела АФ и ОВФ в образующейся в микроканале чипа ТФСЭ.
Время, прошедшее с начала контакта микро-флюидных потоков Ф1 и Ф2, рассчитывали по линейной скорости более медленного потока ИДФ по центральному микроканалу чипа в точке слияния потоков с учетом степени заполнения канала ИДФ. Линейная скорость ИДФ по микроканалу при проведении измерений составляла 4.0 см/с, что приводило к диапазону времен контакта фаз в нем от 0 до 1.12 с.
Величину, равную отношению АС от ОВФ в стационарных условиях к таковой в динамических условиях, считали критерием для оценки равновесности распределения родамина в ТФСЭ. Она составила 93.4 % при С KCl = 300 мМ и 73.7 % без добавки KCl для времени контакта начальных микрофлюидных потоков 1.12 с. Таким образом, солевая добавка способствовала ускорению процессов массообмена в МФЧ. За время контакта первоначальных потоков порядка 1 с процессы массообмена в микроканале чипа при С KCl = = 300 мМ проходили практически до равновесного состояния.
Как видно из рис. 3, б, в, при образовании ТФСЭ в микроканале чипа происходило падение АС в АФ (кривые 2–4 на рис. 3, б; кривые 2, 3 на рис. 3, в) по сравнению с АС от ИДФ в точке слияния начальных фаз (кривая 1 на рис. 3, б, в). АС от АФ для Т конт = 1.12 с составлял не более 35 % от АС этой фазы в равновесных условиях в полностью заполненном микроканале чипа. Наблюдаемое низкое значение АС от AФ в динамических условиях объясняли формированием зоны АФ в микроканале МФЧ толщиной не более 35 мкм при глубине микроканала (110 мкм). Используя эти допущения, из графика 4 (рис. 3, б) можно сделать вывод, что со стороны потока водной фазы (слева) профиль глубины АФ имеет более пологий характер, что связано с механизмом образования ТФСЭ путем перераспределения ацетонитрила в начальной системе фаз.
Отметим, что способом увеличения толщины АФ до высоты микроканала и стабилизации непрерывного "струйного" течения трех фаз в микроканале при меньших скоростях расхода начальных фаз может быть оптимизация топологии микроструктур МФЧ и/или формы поперечного сечения микроканала массообмена, как показано в работах [18, 35].
Быстрота протекания процессов массобмена при образовании ТФСЭ на 1–2 порядка выше по сравнению с проведением жидкостной экстракции в системе двух несмешивающихся жидкостей в МФЧ [6–11]. Это обусловлено прежде всего общими преимуществами всех микрофлю-идных систем (сокращенным временем диффузии вещества за счет уменьшенного расстояния и большим отношением площади межфазной границы к объему фаз), а также выбором растворителей предложенной оригинальной системы с низкими значениями вязкости и механизмом протекания массообмена в условиях одновременного образования новых фаз. Для более детального описания протекающих процессов и их оптимизации с построением физико-химических моделей требуется изучение образования ТФСЭ в микроканале МФЧ при изменении ряда параметров, например составов начальных фаз и температуры.
Тем не менее предложенную на этом этапе изучения систему можно использовать для анализа реальных (био)химических объектов на стадии пробоподготовки и дериватизации, например для определения катехоламинов (КА) в биологических жидкостях (кровь, моча человека) методом ВЭЖХ с флуоресцентным детектированием [24]. Привлекательность использования МФЧ помимо быстроты проведения пробоподготовки будет заключаться в малом расходе биопроб и реагентов в ходе выполнения анализов. При этом квантовый выход флуоресцирующих форм дериватов КА с высокочувствительным групповым реагентом NBD-F при переходе от водной среды к чистому ацетонитрилу и хлористому метилену возрастает в ~3– 7 раз [24].
ВЫВОДЫ
Предложена оригинальная жидкостная система для осуществления экстракции веществ в условиях одновременного с протеканием массообмена образования новой акцепторной фазы. При введении двух исходных фаз в микроканал чипа размером ~200 (Ш)×100 мкм(Г) показана возможность образования трехфазного потока со стабильной поверхностью раздела между всеми фазами. Проведено изучение процесса массообмена в полученной трехфазной системе экстракции (ТФСЭ) на примере распределения родамина 6G между образующимися фазами в стационарных условиях при смешивании двух начальных фаз в стеклянном цилиндре и динамических условиях при контакте двух начальных микрофлюидных потоков в микроканале чипа. Для стационарных условий проведения экстракции родамина измерены аналитические характеристики процесса массообмена (коэффициенты концентрирования и распределения).
Для динамических условий проведения экстракции родамина показана возможность протекания массообмена до равновесного состояния за время около 1 секунды с момента начала контакта мик-рофлюидных потоков при введении добавки хлорида калия в начальную систему фаз. Малое время установления равновесия в микроканале чипа обусловлено диффузией вещества на короткие расстояния в поперечном сечении микроканала, большим отношением площади межфазной границы к объему фаз, а также механизмом протекания массообмена в условиях выделения большей части ацетонитрила из материнского водно-органического раствора в отдельную фазу с одновременным переходом в нее молекул красителя.
