Моделирование белковой структуры α-субъединицы бифенил 2,3-диоксигеназы (BphA1) штамма R. wratislaviensis СН628
 штамма R. wratislaviensis СН628")
Автор: Кирьянова Т.Д.
Журнал: Вестник Пермского университета. Серия: Биология @vestnik-psu-bio
Рубрика: Микробиология
Статья в выпуске: 1, 2025 года.
Бесплатный доступ
Выполнено моделирование трёхмерной структуры α-субъединицы бифенил диоксигеназы (BphA1) штамма Rhodococcus wratislaviensis CH628 с использованием программ MODELLER, AlphaFold и trRosetta. Нуклеотидная последовательность гена bphA установлена при анализе полногеномной последовательности штамма в системе RAST. Филогенетический анализ bphAСН628 показал высокую степень сходства с α-субъединицей нафталин диоксигеназы (narA). Для оценки качества полученных моделей использовались программы ERRAT, VERIFY3D и PROCHECK. Модель BphA1СН628 , построенная с помощью MODELLER, продемонстрировала наивысшую структурную точность, в то время как модель BphA1СН628 AlphaFold лучше предсказала активный центр фермента. Анализ активного центра показал консервативность ключевых аминокислот, участвующих в катализе, что подтверждает функциональную схожесть с нафталин диоксигеназой. Полученные результаты открывают перспективы для дальнейшего исследования BphA1 в контексте его применения в биоремедиации.
Бифенил диоксигеназа, моделирование белков, активный центр
Короткий адрес: https://sciup.org/147251162
IDR: 147251162 | УДК: 579.22/577.29/577.151 | DOI: 10.17072/1994-9952-2025-1-32-42
Modeling of the protein structure of the -subunit of biphenyl 2,3-dioxygenase (BPha1) of the R. Wratislaviensis strain CH628
Three-dimensional modeling of the α-subunit of biphenyl dioxygenase (BphA1) from the Rhodococcus wratislaviensis strain CH628 was performed using MODELLER, AlphaFold, and trRosetta software. The nucleotide sequence of the bphA gene was determined through an analysis of the whole-genome sequence of the strain in the RAST system. Phylogenetic analysis of bphACH628 revealed a high degree of similarity with the α- subunit of naphthalene dioxygenase (narA). To assess the quality of the generated models, ERRAT, VERIFY3D, and PROCHECK programs were employed. The BphA1CH628 model constructed with MODELLER demonstrated the highest structural accuracy, while the BphA1CH628 model from AlphaFold provided a better prediction of the enzyme's active site. Analysis of the active site indicated the conservation of key amino acids involved in catalysis, which supports the functional similarity to naphthalene dioxygenase. These findings open up new avenues for further investigation of BphA1 in the context of its application in the bioremediation.
Текст научной статьи Моделирование белковой структуры α-субъединицы бифенил 2,3-диоксигеназы (BphA1) штамма R. wratislaviensis СН628
Прогнозирование трехмерной структуры белка на основе аминокислотной последовательности является одной из ключевых задач в вычислительной биофизике и молекулярной биологии. Это направление имеет важное значение не только для фундаментальных научных исследований, но и для широкого спектра прикладных задач, таких как интерпретация геномов и прогнозирование функции белков. В последнее время значительный интерес привлекает обратная задача – проектирование последовательностей аминокислот для создания белков с заданной структурой и функциями, что открывает новые перспективы для биотехнологий и медицины [Kuhlman, Bradley, 2019].
Существуют два основных подхода к прогнозированию структуры белков: моделирование на основе шаблона и моделирование без шаблона. Моделирование на основе шаблона использует известные структуры родственных белков для предсказания структуры интересующего белка, тогда как методы без шаблона полагаются на выборку различных конформаций и энергетические оценки для предсказания новых структур. Оба подхода активно развиваются благодаря росту вычислительных мощностей и развитию алгоритмов машинного обучения, что позволяет более точно предсказывать структуры белков и их функциональные области [Jones et al., 2015; Wang et al., 2017].
Одним из важных белков, участвующих в биодеградации токсичных соединений, является бифенил диоксигеназа, ключевой фермент в разложении полихлорированных бифенилов (ПХБ). Эти загрязнители широко распространены в окружающей среде и являются серьезной экологической проблемой из-за их устойчивости к биодеградации и способности накапливаться в живых организмах. В связи с этим изучение структуры и функций бифенилдиоксигеназы является важным направлением исследований, которое может способствовать разработке эффективных методов биоремедиации. В настоящее время построены модели бифенил 2,3-диоксигеназы наиболее известных штаммов-деструкторов ПХБ: BphA1A2 штамма Rhodococcus jostii RHA1 (PDB ID: 1ULI_A), четыре возможных структуры бифенил 2,3-диоксигеназы штамма Burkholderia xenovorans LB400 (PDB ID: 5AEU_A, 2YFI_A, 2XSH_A, 2XR8_A), 3D-структура BphA1 в комплексе с бифенилом штамма Comamonas testosteroni B-356 (PDB ID: 3GZX_A) [Furusawa et al., 2004; Colbert et al., 2013; Dhindwal et al., 2016]. Анализ полученных структур показал, что эффективность связывания фермента с конгенерами ПХБ зависит, в первую очередь, от размера и конфигурации каталитического кармана [Wang et al., 2021]. Среди аминокислот каталитического кармана наибольшее влияние на связывание бифенил 2,3-диоксигеназы с бифенилом оказывают Asp230, Gly335, Asn337, Thr338, Ile339 и Arg340, тогда как в случае, если субстратом является 4,4’-дихлорбифенил, наибольшее влияние оказывают Phe227, Ile336, Asn337, Ile339, Phe378 и Arg340 [Zhu et al., 2020]. Важно отметить, что среди ключевых аминокислот активного центра бифенил 2,3-диоксигеназы при связывании с незамещенным и дизамещенным бифенилом совпадают только 2 позиции: Asn337 и Arg340.
Моделирование структуры α-субъединицы бифенилдиоксигеназы (BphA1) штамма Rhodococcus wratislaviensis CH628 представляет собой важный шаг на пути к созданию эффективных биотехнологических решений для борьбы с загрязнением ПХБ. Этот штамм выделен из почв, длительное время загрязненных хлорорганическими соединениями, и обладает способностью разлагать соединения группы стойких органических загрязнителей, что делает его перспективным для использования в экологических исследованиях и биотехнологиях [Egorova et al., 2017, 2020; Gorbunova et al., 2021].
Цель исследования – создание и анализ моделей α-субъединицы бифенил диоксигеназы (BphA1) штамма Rhodococcus wratislaviensis CH628 с использованием различных программ (MODELLER, AlphaFold и trRosetta).
Материалы и методы исследования
Бактериальный штамм
В работе использован штамм-деструктор полихлорированных бифенилов Rhodococcus wratislaviensis CH628, выделенный ранее из почвы, длительно загрязненной хлорорганическими соединениями [Egorova et al., 2017; Gorbunova et al., 2022]. Штамм CH628 идентифицирован на основании анализа нуклеотидной последовательности гена 16S рРНК (GenBank KX034163) и морфофизиологических признаков.
Построение модели а-субъединицы бифенил 2,3-диоксигеназы (BphAl)
Дедуктивная аминокислотная последовательность BphA1 штамма R. wratislaviensis CН628 получена на основании нуклеотидной последовательности гена bphA (GenBank MW070532), выявленного при анализе полногеномной последовательности штамма с помощью RAST . Нуклеотид- ная последовательность гена bphA была преобразована в аминокислотную последовательность, после чего для обеих последовательностей были построены и визуализированы филогенетические деревья в программе MEGA версии 10.0. Множественное выравнивание последовательностей выполняли с помощью CLUSTALW . Для построения белковой структуры были выбраны программы MODELLER версии 10.4 , AlphaFold и trRosetta .
Поиск шаблонов осуществляли с помощью NCBI Protein BLAST , ограничивая зону поиска базами данных Protein Data Bank (PDB) и UniProt , применяя для выравнивания SWISSMODEL. Для анализа отбирали белковые последовательности с максимальной идентичностью.
Определение качества белковой модели
Для визуализации полученных моделей и предварительного анализа использовали программу PyMOL версии 2.5.4 . С помощью PyMOL оценивали общее качество моделей и проверяли ключевые структурные элементы.
Для оценки качества структурных моделей применяли программы ERRAT [Colovos, Yeates, 1993], VERIFY 3D [Lüthy et al., 1992], PROCHECK , WHATCHECK , ENDscript ipt/ENDscript/).
Выявление области активного центра фермента
Для анализа активного центра фермента использовалась программа Computed Atlas of Surface Topography of proteins (CASTp) [Tian et al., 2018]. CASTp позволяет идентифицировать и характеризовать функционально значимые поверхности и полости белков, включая активные центры. С помощью CASTp были определены размеры и геометрия активного центра BphA1, а также ключевые аминокислоты, участвующие в каталитическом процессе.
Результаты и их обсуждение
Анализ нуклеотидной последовательности гена bphA
Нуклеотидная последовательность гена bphA (GenBank MW070532), кодирующая α-субъединицу бифенил диоксигеназы (BphA1), выявлена при анализе генома штамма R. wratislaviensis CH628 в системе RAST. Анализ нуклеотидной последовательности bphA СН628 выявил высокий уровень сходства с нуклеотидной последовательностью гена narA , кодирующей α-субъединицу нафталин диоксигеназы (NarA) (рис. 1). На рисунке 1 визуализированы эволюционные связи гена bphA СН628 с нуклеотидными последовательностями, уровень сходства с которыми находится в пределах 91,8–100%. В одной «ветке» с исследуемым геном располагаются гены нафталин диоксигеназ штаммов-дестуркторов ароматических соединений, выделенных из района солеотвала горнодобывающего предприятия, очистных сооружений и из почв химического завода [Na et al., 2005; Ananina et al., 2011; Anokhina et al., 2020]. Следует отметить, что такое расположение генов свидетельствует о их высоком сходстве, несмотря на то что данные штаммы выделены с территорий, удаленных друг от друга. Можно предположить, что основным фактором селекции в данном случае выступало химическое загрязнение, а не географическое положение местообитания бактерий.
Анализ первичной структуры BphA1
В результате дедуктивной трансляции с использованием алгоритмов программы MEGA X и сравнения с гомологичными аминокислотными последовательностями, представленными в базе данных GenBank, установлено, что уровень сходства BphA1 СН628 с первичной структурой α-субъединицы нафталин диоксигеназы (КФ 1.14.12.12) штамма-деструктора R. opacus B4 (GenBank BAH47212.1) и α-субъединицы нафталин диоксигеназы штамма Rhodococcus sp. NCIMB12038 (PDB ID: 2B1X|A) составляет 98,6 и 93,7% соответственно. Визуализация математической модели между исследуемой аминокислотной последовательностью и известными показала, что BphA1 СН628 располагается в «ветви» нафталиновых диоксигеназ (рис. 2). При построении филогенетического дерева брали в расчет последовательности, уровень сходства между которыми составлял от 36 до 98%.
9f
>
narAa Rhodococcus ruber PAHs (KY072804)
Rhodococcus sp. PSBB049 plasmid pPSBB049-2 (CP070863) nidA Rhodococcus sp. 124 (AFI21905) narA Rhodococcus sp. DB11 (GQ503239)
■ nahA Rhodococcus sp. WAY 2 plasmid pRWAYOl (CPW6573)
нм
I0O
w
Hi
j Rhodococcus sp. 21391 plasmid pRS02 (CPO63783)
dodA Rhodococcus opacus plasmid pWK30l (ABI 10633)
nidA Rhodococcus opacus o-xylene oxygenase gene cluster (AB206671)
I narAa Rhodococcus sp. GIO (GQ5O3238)
narAa Rhodococcus sp, B13 (GQ5O324O)
hphA R. uratislasiensis (11628
Rhodococcus opacus strain 3D plasmid pLP4!4 (CPI28997)
narA Rhodococcus opacus B4 plasmid pROBOl DNA(APO1II17)
- Rhodococcus rhodochmus strain LH-B3 plasmid unnanied2 (CPI 20358) narA Rhodococcus opacus naphthalene degradation gene cluster (DQ846881)
I лягЛ Rhodococcus sp I BN (AJ4016I2)
Rhodococcus opacus strain R7 plasmid pPDG4 (CPOO895D rnoA Rhodococcus sp. CIR2 (AB024936)
narAa Rhodococcus sp. NC1MB12038 (АЮ82663)
Rhodococcus oxyhenzoniwrans strain S2-17 plasmid pRB98 ((’14)21355)
nahA Rhodococcus sp, WAY2 plasmid pRWAYOl (CP046573)
Rhodococcus opacus strain Cl plasmid pCl 3 (CP137574)
Rhodococcus opacus strain S8 plasmid pLPS8 (CP093381)
Rhodococcus opacus strain 9 (CP095405)
0.25 0.20 0.15 0.10 0.05 0.00
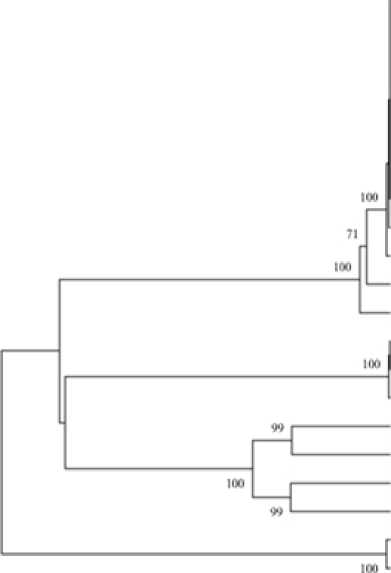
Рис. 1. Положение гена bphA R. wratislaviensis CH628 на филогенетическом дереве, построенного на основании анализа нуклеотидных последовательностей с использованием метода UPGMA.
Цифрами показана достоверность ветвления, установленная с помощью “butstrap”-анализа
[Position of the bphA gene of R. wratislaviensis CH628 on the phylogenetic tree constructed based on the analysis of nucleotide sequences using the UPGMA method.
The numbers show the reliability of branching established using the bootstrap analysis]
Rieske Rhodococcus opacus (Nocardia «раса) (UniProt AOA848PLK7)
NirA Rhodococcus opacus (Nocardia opacai (UniProt Q2WG94)
NarA Rhodococcus opacus «train 154 (UniProt|ClBE09)
NarA Rhodococcus sp. BI3 (UniProt F4Y5R3)
NirA Rhodococcus sp. G10 (UniProt G3BJU7)
Rhodococcus opacus (AlphaFold|Q76BV8)
NarA Rhodococcus imtechensis RKJ300 (JCM 13270) (UniProt|lOWRI2)
Rhodococcus opacus (Nocardia opaca) (UniProt Q76BV8)
Rhodococcus sp. S2-I7 (UniProt AOA2S2C575)
NahA Rhodococcus sp. WAY2 (UniProt A0A6PI1EU3)
BphA R. wratislaviensis CH628
NarA Rhodococcus sp. NCIMB 12*38 (pdb 2BIX)
NarA Pseudomonas putida (pdb I F.G9 I)
NarA Pseudomonas sp. С18 (pdb 41IJL I)
NarA Pseudomonas sp. (pdb 2HMJ I)
BphA Rhodococcus jostii RHAI (pdb|lULI)
BcnA Pseudomonas putida (pdb 3ENI)
BphA Hurkholderia xenmorans L15400 (pdb|2XR8)
BphA Comamonas testostemni (pdb|3GZX)
Ricskc Comamonas testosteroni KF-I (pdb7VJU)
TphA Comamonas sp. Gallus gallus (pdb 7QO5 2)
4------------------1-----------------1-----------------1
030 020 0.10 0.00
Рис. 2. Положение BphA1 штамма R. wratislaviensis CH628 на филогенетическом дереве, построенном на основании сравнительного анализа аминокислотных последовательностей с использованием метода UPGMA.
Цифрами показана достоверность ветвления, установленная с помощью “butstrap”-анализа
[Position of BphA1 of the R. wratislaviensis CH628 strain on the phylogenetic tree constructed based on the comparative analysis of amino acid sequences using the UPGMA method.
The numbers show the reliability of branching established using the bootstrap analysis]
Моделирование вторичной и третичной структуры BphA1
Вторичная структура α-субъединицы бифенил диоксигеназы (BphA1) штамма
R. wratislaviensis CH628 была получена с использованием программ MODELLER, AlphaFold и trRosetta, отличающихся алгоритмами построения белковой структуры.
В основе моделирования вторичной и третичной структур белков в программе MODELLER лежит принцип построения по известной модели белка, характеризующейся наибольшим уровнем сходства первичной структуры с аминокислотной последовательностью исследуемого белка/фермента. В рамках настоящего исследования в качестве модели была выбрана классическая структура NarA1, описанная у штамма Rhodococcus sp. NCIMB12038 (PDB ID: 2B1X|A), которая показала высокий уровень сходства с анализируемой последовательностью (идентичность 93.72% для аминокислотной последовательности). В результате использования алгоритмов программы MODELLER на основе NarA1 NCIMB12038 было построено пять моделей BphA1 штамма R. wratislaviensis CH628, отличающихся по показателю энергии. Для дальнейшего исследования была выбрана модель BphA1 CH628 с наименьшим показателем DOPE (-45531.87).
Построение моделей белков без шаблона возможно при использовании программ AlphaFold и trRosetta. Однако данные программы используют различные подходы для прогнозирования новой структуры. В AlphaFold используются алгоритмы мономерной модели, тогда как в trRosetta используются нейронные сети глубокого обучения и алгоритм Rosetta. Использование данных программных комплексов позволило получить две модели BphA1 CH628 . Параметры вторичной структуры α-субъединицы бифенил диоксигеназы штамма R. wratislaviensis CH628 представлены в табл. 1. Для удобства описания модели BphA1, полученные с применением различных программ обозначены как:
BphA1(М) – построена в программе MODELLER,
BphA1(AF) – разработана с применением AlphaFold,
BphA1(TR) – смоделирована с использованием базовых параметров trRosetta.
Таблица 1
Характеристики вторичной и третичной структуры B ph A1сн628 [Characteristics of the secondary and tertiary structure of BphA1 СН628 ]
|
Элемент структуры |
BphA1(М) |
BphA1(AF) |
BphA1(TR) |
|
α спирали |
11 |
13 |
13 |
|
π-спирали |
5 |
5 |
6 |
|
строгие α-витки |
2 |
3 |
3 |
|
β складчатости |
20 |
20 |
17 |
|
Строгие β-витки |
10 |
10 |
12 |
|
Объём Å 3 |
49,40 |
50,41 |
48,60 |
|
Площадь Å 2 |
18,66 |
19,26 |
21,01 |
|
Количество АК |
401 |
401 |
401 |
Использование алгоритма построения «по шаблону» (MODELLER) привело к созданию модели BphA1(М), отличающейся меньшим количеством α спиралей и наименьшей площадью молекулы, тогда как объем характеризуется средним значением между показателями объема для BphA1(AF) и BphA1(TR). Модель BphA1(TR) обладает наименьшим объемом при наибольшей площади молекулы, содержит большее количество π-спиралей и строгих β-витков.
3D-Модели BphA1 CH628 были визуализированы и предварительно проанализированы с использованием программы PyMOL (рис. 3а–в). Для наглядного сравнения 3D-модели BphA1(М), BphA1(AF) и BphA1(TR) были выровнены относительно друг друга (рис. 3г), что позволило оценить различия в структурных элементах.
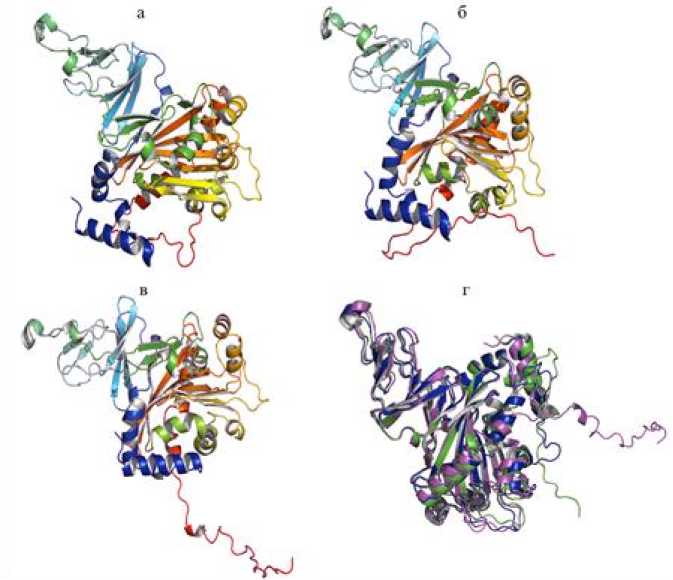
Рис. 3. 3D-Модель BphA1 CH628 : а – BphA1(М), б – BphA1(AF), в – BphA1(TR), г – выровненные относительно друг друга модели BphA1(М) (синий), BphA1(AF) (зелёный) и BphA1(TR) (розовый) [3D-Model of BphA1CH628: a – BphA1(M), b – BphA1(AF), c – BphA1(TR), d – aligned relative to each other models of BphA1(M) (blue), BphA1(AF) (green) and BphA1(TR) (pink) ]
Из рисунка 3г видно, что петлевые области BphA1CH628, особенно на периферии, имеют заметные различия между моделями. Модель BphA1(TR) демонстрирует больше отклонений от структуры BphA1(М) и BphA1(AF) в вытянутых петлях. По всей видимости, trRosetta с меньшей точностью моделирует гибкие области белка, чем MODELLER и AlphaFold. Можно предположить, что BphA1(TR) характеризуется менее стабильной структурой, т. к. содержит менее компактные петлевые области. Данное предположение подтвердилось при оценке качества моделей.
Качество полученных моделей BphA1 CH628 оценивали с использованием нескольких независимых методик анализа структурных моделей, включая ERRAT, VERIFY3D и PROCHECK (табл. 2, рис. 4).
Таблица 2
Сравнение параметров качества полученных моделей белка [Comparison of the quality parameters of the obtained protein models]
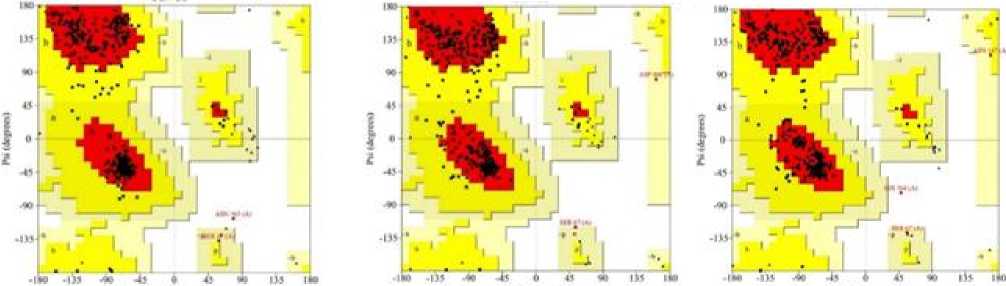
ЛиЛ^т, ЖЩИЧ П»М*>аМ
Рис. 4. График Рамачандрана модели BphA1 CH628 , построенный с использованием (а) Modeller10.4, (б) AlphaFold, (в) TrRosetta
[Ramachandran plot of BphA1CH628 model built using (a) Modeller10.4, (б) AlphaFold, (в) TrRosetta ]
BphA1(М) показала наивысшую точность: 90.7% аминокислотных остатков находились в благоприятных регионах графика Рамачандрана, а ERRAT-показатель составил 87.53%. BphA1(AF) продемонстрировала высокую точность в предсказании активного центра фермента, хотя её общие показатели ERRAT (90.8%) и VERIFY3D (82.79%) были чуть ниже по сравнению с BphA1(М). Модель BphA1 CH628 , созданная с помощью trRosetta, оказалась наименее точной, так как ее показатель VERIFY3D составил 71.82%. Таким образом, BphA1(М) характеризуется наиболее точной структурой.
Построенные модели служат основой для дальнейшего структурного и функционального анализа, позволяя сравнить BphA1 с другими диоксигеназами и выявить уникальные или консервативные структурные особенности.
В рамках настоящего исследования было осуществлено сравнение модели BphA1 CH628, полученной с использованием MODELLER с моделями α-субъединиц известных штаммов-деструкторов ароматических соединений. Анализ показал, что BphA1CH628 структурно ближе к α-субъединице нафталин диокси- геназы штаммов Rhodococcus sp. NCIMB12038 и Rhodococcus opacus B4. Для сравниваемых аминокислотных последовательностей характерны длинные области с одинаковым набором аминокислот. Сравнение количественных показателей вторичной структуры BphA1CH628 c NarANCIMB12038 и NarAB4 выявило различие в одну α-спираль, 2-3 η-спирали, и одну β-складчатость.
Принципиально другую картину мы получили при сравнении BphA1 CH628 с α-субъединицей бифенил диоксигеназы штамма Rhodococcus jostii RHA1 (рис. 4б). Выявлено, что при незначительных отличиях в количественных показателях расположение спиралей и складчатостей практически не совпадало.
Полученные результаты позволяют предположить, что бифенил диоксигеназа штамма R. wrati-slaviensis CH628 способна катализировать реакции, характерные для нафталиновых диоксигеназ.
Анализ активного центра BphA1 штамма R. wratislaviensis CH628
Анализ активного центра фермента BphA1 CH628 был выполнен с использованием программы CASTp. Установлено, что модель BphA1(AF), созданная в программе AlphaFold, наиболее точно предсказала расположение активного центра (рис. 5).
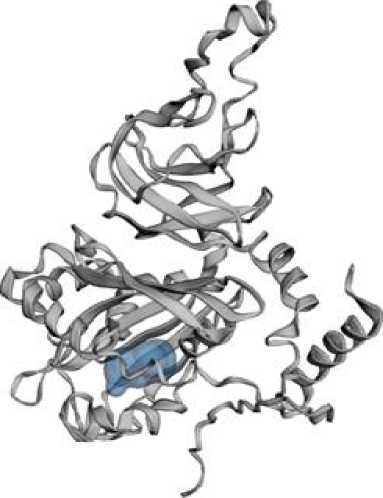
Рис. 5. Модель ВphA1 R. wratislaviensis CH628, полученная при помощи системы искусственного интеллекта AlphaFold. Синим цветом выделена предполагаемая область активного центра
[Model of BphA1 of R. wratislaviensis CH628 obtained using the AlphaFold artificial intelligence system. The putative active site region is highlighted in blue ]
На основании полученных данных можно предположить, что субстрат-связывающий карман в каталитическом домене α-субъединицы BphA1 СН628 формируют спирали α5, α6, α7, α10, α11, β-нити β15, β16, β17, петли α10–α11 и β14. В активном центре находятся аминокислоты Asn209, Phe210, Asp213, Ala214, His216, Thr217, His221, Met 224, Ala230, Phe236, Ala237, Ile254, Phe293, His295, Phe307, Met309, Thr361, Leu362, Ala369. Следует отметить, что аминокислоты His216 и His221 также присутствуют в составе активного центра нафталин диоксигеназы штамма Rhodococcus sp. NCIMB12038 (PDB ID: 2B1X|A) и участвуют во взаимодействии с ионом Fe³⁺.
У штамма Rhodococcus jostii RHA1 карман связывания субстрата расположен между основным β-слоем и α-спиралями в каталитическом домене α-субъединицы бифенил диоксигеназы и формируется спиралями α6, α7, α8, α9, β-нитью 16, петлями α13–α14 и β17–β18, включая остатки вокруг иона Fe³⁺ (Gln217, Phe218, His224, His230 и Asp378). В кармане связывания субстрата два кольца связанной молекулы бифенила расположены не параллельно, а со смещением. Каждое кольцо субстрата зафиксировано с обеих сторон; кольцо 1 располагается между His224 и Leu323, а кольцо 2 – между Ala225 и Phe368 [Furusawa et al., 2004].
Сравнение активных центров BphA1CH628 и BphA1RHA1 показало, что аминокислоты Ala225 и Leu323, взаимодействующие с молекулой бифенила в активном центре штамма R. jostii RHA1 и находящиеся в таких же позициях Val225 и Leu323 штамма R. wratislaviensis CH628 (рис. 4б) относятся к группе нейтральных гидрофобных аминокислот, что может обеспечивать взаимодействие активного центра BphA1CH628 с молекулой бифенила. Одной из аминокислот, участвующих во взаимодействии с ионом Fe³⁺, в BphA1RHA1 является Gln217. В данной позиции в составе BphA1CH628 находится Thr217 (рис. 4б). Поскольку данные аминокислоты принадлежат группе полярных гидрофильных аминокислот, можно предположить, что Thr217 активного центра BphA1CH628 также участвует во взаимодействии с ионом Fe³⁺.
Сходство ряда ключевых аминокислотных остатков активного центра BphA1 CH628, NarA NCIMB12038 и BphA1 RHA1 позволяет предположить, что бифенил диоксигеназа штамма R. wratislaviensis CH628 способна эффективно катализировать диоксигеназные реакции, необходимые для разложения как нафтали-на/полициклических ароматических углеводородов, так и полихлорированных бифенилов.
Заключение
В ходе данного исследования были созданы и проанализированы три модели бифенил 2,3-диоксигеназы (BphA1) штамма R. wratislaviensis CH628 с использованием программ MODELLER, AlphaFold и trRosetta. Каждая из моделей прошла тщательную оценку качества с помощью таких инструментов, как ERRAT, VERIFY3D и PROCHECK. Наиболее высокие показатели точности структуры продемонстрировала модель, созданная с помощью MODELLER, где более 90% аминокислотных остатков находятся в благоприятных регионах, что делает её наиболее достоверной для общего структурного анализа. Однако модель AlphaFold показала наибольшую точность в предсказании активного центра фермента.
Анализ активного центра показал значительное сходство между BphA1 штамма R. wratislaviensis CH628 и нафталин диоксигеназой (NarA1) штамма Rhodococcus sp. NCIMB12038. Большинство консервативных аминокислотных остатков, таких как His216 и His221, которые участвуют во взаимодействии с субстратами и ионами металлов, присутствуют в обеих последовательностях. Также выявлены сходства в строении субстрат-связывающего каталитического кармана BphA1 CH628 и BphA1 RHA1 и некоторых ключевых аминокислотных остатков активного центра.
Таким образом, модель BphA1 CH628 , построенная с помощью AlphaFold, наиболее пригодна для дальнейших исследований, связанных с функциональным анализом активного центра фермента, в то время как модель BphA1 CH628 , созданная в программе MODELLER, рекомендуется для общего структурного анализа и понимания стабильности белка. Полученные данные открывают перспективы для дальнейшего структурного и биохимического изучения бифенил диоксигеназы штамма R. wratislaviensis CH628.
