Моделирование и анализ виртуального растяжения эластичного и пластичного кристаллов дигалогенфенолов

Автор: Решетникова Регина Викторовна, Матвейчук Юрий Васильевич, Барташевич Екатерина Владимировна
Журнал: Вестник Южно-Уральского государственного университета. Серия: Химия @vestnik-susu-chemistry
Рубрика: Физическая химия
Статья в выпуске: 3 т.14, 2022 года.
Бесплатный доступ
С целью проверки устойчивости результатов моделирования растягивающих деформаций молекулярных кристаллов проведен тест на виртуальное растяжение пластичного 3,4-дихлорфенола, эластичного 4-бром-3-хлорфенола. Моделирование всех рассматриваемых кристаллических структур было проведено методами теории функционала плотности с учетом периодических граничных условий и локализованных базисных наборов. Подтверждено, что предложенный нами ранее виртуальный тест на растяжение молекулярных кристаллов позволяет объяснять и предсказывать их механические свойства по характерному поведению изменений энергии кристаллической ячейки и ее объема в зависимости от растягивающих деформаций. В данной работе растягивающие деформации кристаллической ячейки 3,4-дихлорфенола проводили с разным шагом в 1 и в 3 % от величин параметров ячейки. Это позволило нам установить, что тренды изменения кристаллической структуры и рассматриваемых свойств мало зависят от величины шага растяжения в изученных пределах. Были обнаружены лишь небольшие отличия в энергии кристаллической ячейки на 15 % растяжения, которые позже нивелировались. Для изученных структур кристаллов выявлено, что четыре водородные связи O-H…O прочнее удерживают сформировавшиеся синтоны из четырех молекул, чем межстопочные взаимодействия Cl…Cl или Br…Br, которые способны к переключению или разрыву в процессе деформации. При этом оказалось, что взаимодействие галогенов I типа как для эластичного, так и для пластичного кристалла теряется в ходе растяжения, при этом вместо одного ван-дер-ваальсового взаимодействия формируются две галогенные связи Cl…Br и Сl…Cl, соответственно. В зависимости от растягивающих деформаций на начальном этапе растяжений обнаруживаются сходные изменения в поведении структуры, энергии кристаллической ячейки и ее объема как для пластичного 3,4-дихлорфенола, так и для эластичного 4-бром-3-хлорфенола. Тем не менее пластичный 3,4-дихлорфенол отличается более медленными изменениями в структуре и коротким периодом замедления роста энергии кристаллической ячейки с появлением межстопочных зазоров. Для эластичного кристалла этот период значительно более протяженный и сопровождается осцилляциями в изменении энергии. Таким образом, замена атома Cl на Br в 4-м положении фенольного цикла приводит к тому, что при определенном проценте растягивающих деформаций формируются достаточно большие полости, что способствует существенным изменениям в структуре без значительных затрат энергии.
Виртуальный тест на растяжение, молекулярные кристаллы, квантово-химические расчеты с периодическими граничными условиями, механические свойства, водородные связи, галогенные связи
Короткий адрес: https://sciup.org/147238626
IDR: 147238626 | УДК: 544.162.2 | DOI: 10.14529/chem220313
Simulation and analysis of virtual tensile of elastic and plastic crystals of dihalophenols
In order to check the stability of tensile deformation modeling results of molecular crystals, a test for virtual stretching of plastic 3,4-dichlorophenol and elastic 4-bromo-3-chlorophenol has been carried out. Modeling for all considered crystal structures has been carried out by density functional theory methods with allowance for periodic boundary conditions and localized basis sets. It has been confirmed that the previously proposed virtual tensile test of molecular crystals makes it possible to explain and predict their mechanical properties from the characteristic behavior of changes in the energy of a crystal cell and its volume depending on tensile strains. In the present study the tensile deformations of the crystal 3,4-dichlorophenol cell have been modeled with different steps (1% and 3%) of the values of the cell parameters. This has allowed us to establish that the trends in the crystal structure change and properties under consideration depend but little on the value of the stretching step within the studied limits. Only small differences in the crystal cell energy per 15% stretching have been found; they have later leveled out. For the studied crystal structures, it has been found that four O-H…O hydrogen bonds hold the formed synthons of four molecules with greater strength than the Cl…Cl or Br…Br interstack interactions, which are capable of switching or breaking during deformation. It has turned out that the interaction of type I halogens for both elastic and plastic crystals is lost during stretching, and instead of one van der Waals interaction, two halogen bonds, Cl…Br or Сl…Cl, respectively, are formed. Depending on the tensile strains at the initial stage of stretching, similar changes in the behavior of the structure and energy of a crystal cell, as well as its volume are discovered for both plastic 3,4-dichlorophenol and elastic 4-bromo-3-chlorophenol. However, plastic 3,4-dichlorophenol is characterized by slower changes in the structure and a shorter period of deceleration of the crystal cell energy growth with the appearance of interstack gaps. For an elastic crystal this period is much longer and is accompanied by “oscillations” in the energy change. Thus, the replacement of the chlorine atom by bromine in the 4th position of the phenol cycle leads to formation of reasonably large cavities if tensile strains reach a certain percentage, which contributes to significant changes in the structure without significant energy input.
Текст научной статьи Моделирование и анализ виртуального растяжения эластичного и пластичного кристаллов дигалогенфенолов
Квантово-механическое моделирование с учетом периодических граничных условий, основанное на теории функционала плотности (DFT) [1, 2], представляет собой эффективное средство для предсказания и характеристики свойств кристаллических структур как в атомистическом масштабе, так и на уровне свойств электронной плотности. Детальное химическое и физическое понимание, обеспечиваемое первопринципным теоретическим моделированием, достигается с помощью квантово-химических расчетов структуры и свойств кристаллов, которые уже можно относить к материалам. Вычислительный подход является стандартным инструментом для экспериментальной проверки, интерпретации и прогнозирования во многих областях исследований [3, 4]. В настоящее время существует потребность в новых методах прогнозирования механических свойств синтезируемых кристаллических материалов для фармацевтической промышленности. К этому располагает как накопленный практический опыт, так и подходы, основанные на варьировании заместителей в биологически активных соединениях для получения новых более эффективных и менее токсичных лекарств. Однако это может повлиять на механические свойства кристаллических материалов, что важно учитывать при разработке технологических этапов таблетирования и измельчения [5].
Имеющиеся компьютерные исследования в основном полагаются на изучение влияния свойств добавок путем моделирования методом Монте-Карло [6–8]. Часть исследований посвящена исследованию физических свойств и фазовых переходов методами классической молекулярной динамики [9], при анализе работ, посвященных деформациям в кристаллах, можно заметить, что большинство работ посвящено исследованию деформаций под нагрузкой [10–12], в то время как работ по растягивающим деформациям крайне мало [13]. Мотивацией нашего исследования является заполнение пробела в области знания о том, что происходит с кристаллами на уровне укладки отдельных молекул и химических связей, и как при этом изменяются их свойства. Полученные результаты позволят в будущем создавать материалы с заранее заданными свойствами, что имеет большое значение для их применения в медицине и технике.
Существенной особенностью кристаллических тел является их структурная анизотропия, которая приводит к зависимости ряда свойств от направления в кристалле. Кроме того, известно, что многие свойства кристаллов определяются их структурной организацией, которая, в свою очередь, может включать дефекты. Поэтому большой интерес представляют знания о деформации кристаллов и отклик структуры на механические воздействия, а именно, изучение нековалентных взаимодействий и эффектов, вызываемых растягивающими деформациями, что позволит прогнозировать влияние структурных дефектов в кристаллах на свойства состоящих из них материалов. Определенный интерес вызывает поведение галогенных связей [14, 15] и их сравнение с водородными связями в контексте структурной химии. Это важно для развития инженерии и проектировании материалов [16], поскольку совместное присутствие, конкурентное и синергетическое поведение водородных и галогенных связей оказывает сильное влияние на свойство кристалла, подвергающийся деформации.
В нашей работе нам было важно понять, каким образом ведут себя хрупкие, пластичные и эластичные кристаллы при растягивающих деформациях и возможно ли прогнозирование этих свойств с помощью теста на растягивающие деформации. Также мы ставили задачу сравнить поведение водородных и галогенных связей при растягивающих деформациях.
Методы расчетов
Моделирование всех рассматриваемых кристаллических структур было проведено методом Кона-Шэма (DFT) с периодическими граничными условиями при использовании пакета программ CRYSTAL17 (v.1.0.2) [17, 18]. Начальные данные (пространственная группа симметрии, параметры элементарной ячейки и координаты атомов) взяты из Кембриджской кристаллографической базы данных [19]. Для расчётов был использован скорректированный метод Хартри – Фока HF-3c [20]. Этот метод включает в себя базисный набор MINIX [21] и три полуклассиче-ские поправки: дисперсионная поправка Гримме D3 [22], атомное попарное приближение коррекции противовеса gCP [23, 24] и ближняя поправка базиса SRB. Для каждой структуры предварительно проводилась полная оптимизация параметров ячейки и координат атомов (опция FULLOPT) с сохранением пространственной группы, то есть поиск минимума на поверхности потенциальной энергии с полной релаксацией кристаллической структуры. При этом использовались следующие параметры сходимости: по энергии в SCF-цикле 10–10 aт. ед., по градиенту 10–5 aт. ед. и по смещению 3·10–5 aт. ед.
Виртуальный тест растяжения кристаллических структур дигалогенфенолов проводился согласно алгоритму, предложенному в работе [13]. Для фиксации определённой деформации кристалла по осям a, b и c, по-отдельности для каждой из них, применялись опции CRYDEF и FIXDEF программы CRYSTAL17, с сохранением пространственной группы и всех операторов симметрии. Эти опции позволяют проводить оптимизацию координат атомов и параметров кристаллической ячейки, когда один из этих параметров сохраняется постоянным во время оптимизации. Чтобы смоделировать постоянную деформацию растяжения, мы взяли предварительно полностью оптимизированную, равновесную в заданных условиях расчёта, кристаллическую структуру, увеличили один из параметров ячейки на 3 % и зафиксировали его при оптимизации новой, растянутой структуры. После того, как остальные параметры ячейки и координаты атомов были вновь оптимизированы, величина этого параметра увеличивалась ещё на 3 % относительно начального значения (в равновесной структуре), фиксировалась, и далее проводилась новая оптимизация. Такую пошаговую оптимизацию с растяжением кристалла по одной из осей мы повторяли десять раз, до 30 % удлинения кристалла по выбранной оси.
В аналогичных условиях был проведён повторный тест растяжения с уменьшенным до 1 % шагом увеличения каждого параметра ячейки. Это было сделано для выяснения, зависят ли тренды изменения структуры и её рассматриваемых свойств от величины шага, то есть от «плавности» или «равномерности» последовательного увеличения растягивающей деформации. Следует отметить, что в силу симметрии для кристаллических структур 3,4-дихлорфенола и 4-бром-3-хлорфенола, кристаллизующихся в тетрагональной пространственной группе, растяжение проводилось одновременно по осям а и b . Фактически, кристалл растягивался изотропно в любом направлении, параллельном плоскости ab . Далее, после выполнения расчётов, были построены зависимости изменения объёма и полной энергии кристаллографической ячейки относительно величин исходной оптимизированной структуры от процента увеличения каждого параметра ячейки и проанализированы изменения конфигурации связей и структурных особенностей полученных кристаллических форм.
Результаты и обсуждение
Нами были проанализированы результаты моделирования структуры и растягивающих деформаций пластичного 3,4-дихлорфенола (I 4 1 /a), эластичного 4-бром-3-хлорфенола (I 4 1 /a) (рис. 1). Эти кристаллы ранее были исследованы экспериментально в работе [25] на предмет сопоставления их механических свойств с особенностями межмолекулярных взаимодействий.
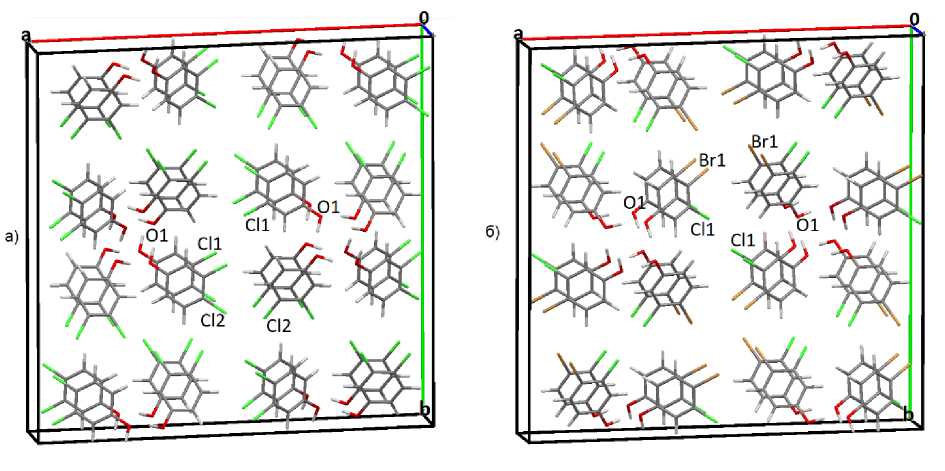
Рис. 1. Расположение молекул, входящих в состав кристаллов: а – 3,4-дихлорфенола, б – 4-бром-3-хлорфенола
В кристалле сформированы стопки из синтонов, сформированных четверками молекул, связанных в слое водородными связями OI...H4, между кислородом гидроксильной группы одной молекулы и водородом соседней молекулы. Следует сказать, что эти водородные связи повторяются вдоль оси c , и четыре связи образуют виток спирали в стопке высотой в 3,691 Å. При одновременном растяжении по кристаллографическим осям a и b одна из молекул приподнимается и встраивается между молекулами соседнего слоя. При этой деформации синтоны не разрушаются, но молекулы в стопках немного поворачиваются относительно друг друга.
Интересна смена типа нековалентного взаимодействия галоген-галоген: с типа I - ван-дер-ваальсового взаимодействия, при котором а-дырка одного хлора ориентирована с небольшим смещением на σ-дырку другого хлора, на тип II – галогенную связь, при которой σ-дырка ориентируется на неподеленную пару электронов (рис. 2). При 15 % растяжении имеется взаимодействие Сl1…Cl1 с длиной 3,553 Å, соответствующее типу I, которое при растяжении уменьшает свою длину, хотя связь Cl1…Cl1 все ещё остается I типа. При 18% растяжении происходит «переключение» взаимодействий, при котором вместо одного контакта Cl1…Cl1 возникают две галогенные связи Cl1…Cl2, причем они становятся более длинными, чем предшествующее взаимодействие типа I.
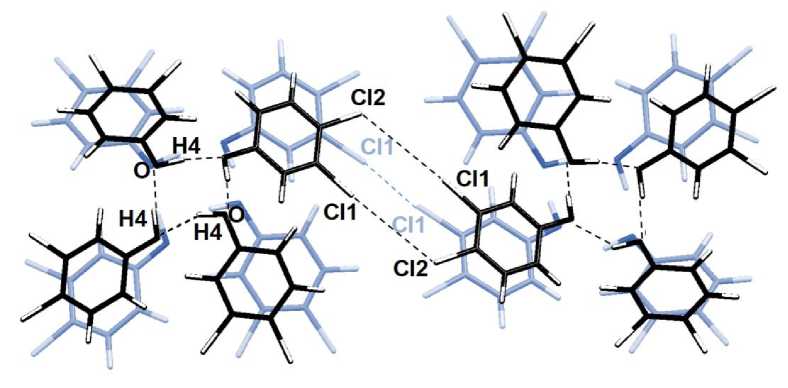
Рис. 2. «Переключение» в кристалле 3,4-дихлорфенола нековалентной связи Cl…Cl с типа I (ван-дер-ваальсового взаимодействия) на тип II (галогенную связь)
При растяжении по оси c происходит заметное укорочение контакта Cl1…Cl1 с 3,257 Å в равновесной форме до 3,236 Å при 18 % растяжении с дальнейшим ростом до 3,250 Å при максимальном растяжении. При этом расстояние между соседними слоями, которое можно оценить по расстоянию между атомами Cl1 и H2, равному 3,045 Å в равновесной структуре, уменьшается при 30 % растяжении – указанное расстояние составляет 2,972 Å. Это свидетельствует о сближении молекул по оси с за счет их разворота в одной из стопок. Расстояние между атомами Cl2 и H2, наоборот, растет: с 3,139 до 3,216 Å соответственно. Таким образом, при растяжении по оси с изменение межслоевых расстояний проявляется заметно, но при этом не появляется никаких полостей или трещин.
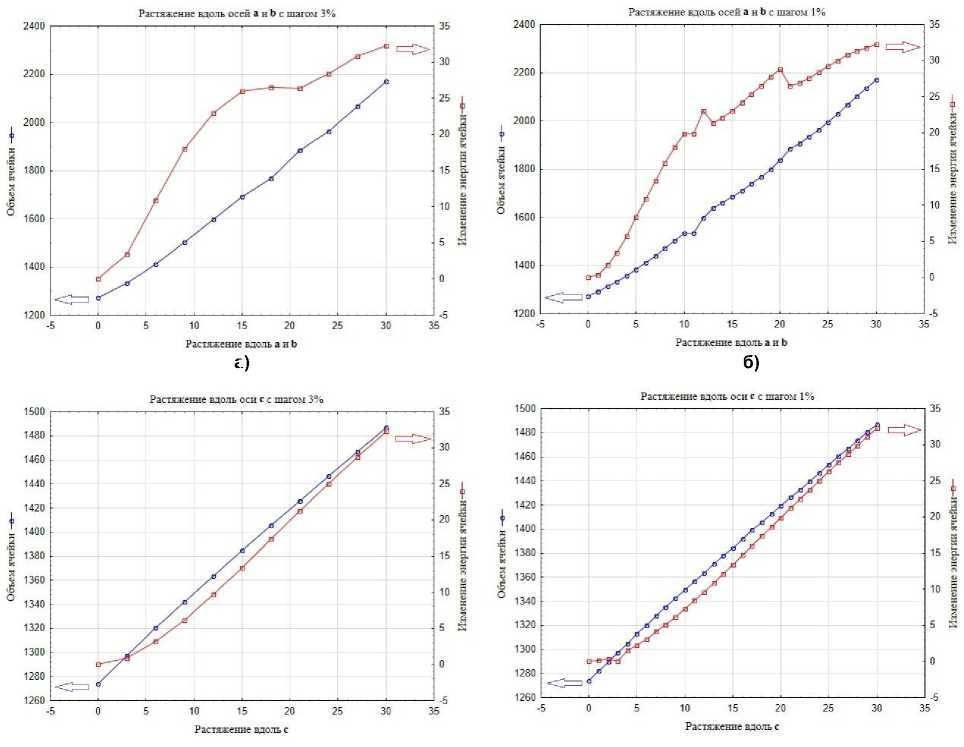
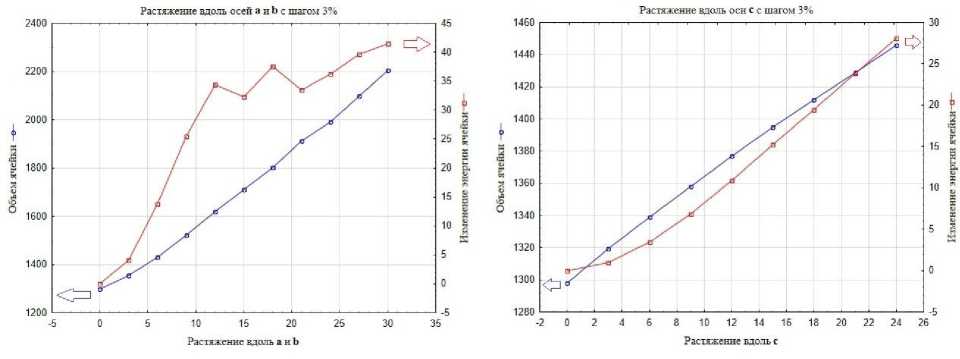
При рассмотрении графиков на рис. 3а–б видно, что изменение энергии ячейки ΔE перестает резко расти и выходит на небольшое плато, начиная с 15 % растяжения по осям а и b . Это наблюдение можно объяснить, во-первых, «переключением» типа нековалентной связи Cl1…Cl1, описанным выше, во-вторых, появлением зазоров в двух ортогональных направлениях. При рассмотрении графика растяжения кристалла по оси с (рис. 3в-г), выхода на плато не наблюдается.
Из сравнения графиков на рис. 3а-г, демонстрирующих зависимости объёма и изменения полной энергии кристаллографической ячейки 3,4-дихлорфенола от процента удлинения каждого параметра ячейки, следует, что выбор шага не повлиял на поведение кристаллической структуры при ее растяжении. Изменения энергии и объема ячейки происходят симбатно в обоих режимах растяжения. При растяжении с шагом в 1 и 3 % относительно исходной структуры не появляется значимых различий как в достигнутом объеме кристаллографической ячейки, так и в увеличении энергии ячейки. При сравнении нековалентных взаимодействий и межатомных расстояний, получаемых на одинаковом проценте растяжения, можно заметить, что самая большая разница наблюдается для взаимодействия Cl1…Cl1 на 12 % растяжения, и составляет всего 0,022 Å. В этой области мы видим не только изменение энергии, но и небольшие различия в относительных ориентациях молекул. При дальнейших шагах растяжения такое расхождение нивелируется. Следует также отметить, что в режиме растяжения с малым шагом в 1 % изменение энергии ячейки при одинаковых растяжениях оказывается немного меньше, чем при растяжении с большим шагом в 3 %, что видно на рис. 4.
а)
в)
г)
Рис. 3. Зависимости объёма и изменения полной энергии кристаллической ячейки относительно исходной оптимизированной структуры от процента удлинения каждого параметра ячейки для 3,4-дихлорфенола: а – при растяжении вдоль осей a и b с шагом в 3 %; б – при растяжении вдоль осей a и b с шагом в 1 %; в – при растяжении вдоль оси c с шагом в 3 %; г – при растяжении вдоль оси c с шагом в 1 %
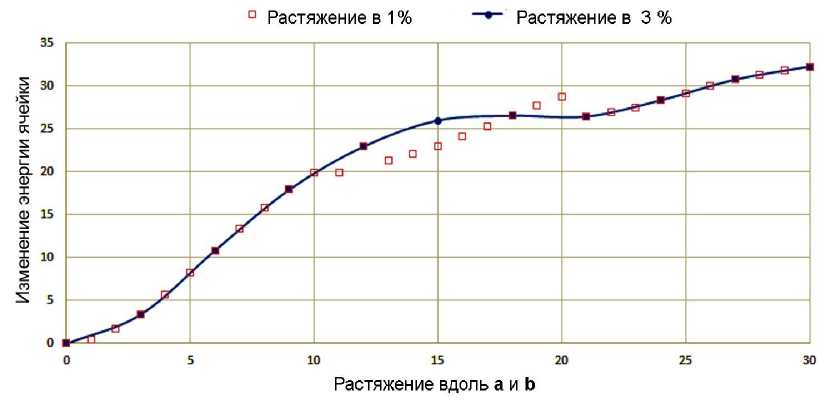
Рис. 4. Разница в изменении энергии кристаллической ячейки при виртуальном растяжении с разным шагом 1 и 3 % кристалла 3,4-дихлорфенола
Эластичный кристалл 4-бром-3-хлорфенола имеет сходную структуру кристаллических мотивов, состоящих из синтонов, сформированных четырьмя ближайшими молекулами, связанными водородными связями (рис. 5). Эти четверки водородно-связанных молекул имеют тенденцию формировать стопки вдоль оси с , которые сохраняются при растягивающих деформациях. При одновременном растяжении вдоль осей а и b с шагом 3 %, при 12 % удлинении появляются кластеры, состоящие из отделяющихся друг от друга стопок. Этот процесс сопровождается началом формирования полостей, возникающих при удлинении межатомных расстояний. Следует заметить, что галогенные связи Br1…Br1 при этом утрачиваются, позволяя сформироваться пустотам между стопками.
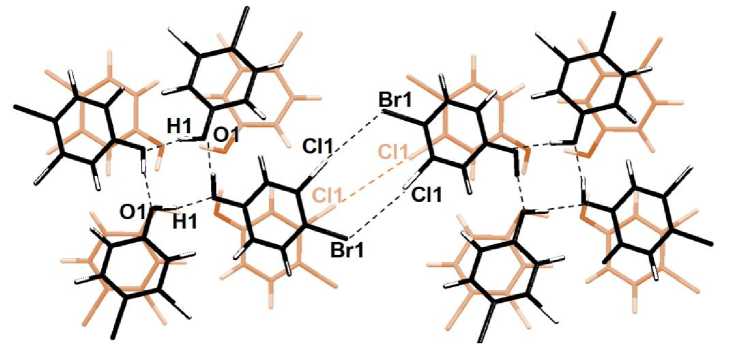
Рис. 5. «Переключение» в кристалле 4-бром-3-хлорфенола нековалентной связи Cl…Cl с типа I (ван-дер-ваальсового взаимодействия) на тип II (галогенную связь Cl…Br)
Из-за появляющихся полостей в кристаллической упаковке возникает возможность существенного синхронного поворота всех четырех молекул в стопке. В результате, на 15 % растяжении происходит смена типа структурообразующих нековалентных взаимодействий, реализуемых посредством галогенов: взаимодействие типа I Cl…Cl сменяется парными галогенными связями Сl…Br и Br…Cl, формирующимися между соседними фенольными циклами.
При растяжении кристалла 4-бром-3-хлорфенола по оси с формирования полостей не наблюдается. Поведение нековалентных взаимодействий между молекулами сильно отличается: с растяжением межстопочное взаимодействие Cl1…Cl1 становится короче, при этом галогенная связь Br1…Br1 не теряется, но становится длиннее.
При рассмотрении графика (рис. 6а), видно, что изменение энергии ячейки ΔE перестает резко расти и осциллирует, начиная с 12 % растяжения по осям а и b . Это явление сопровождает тенденцию к возникновению полостей. При рассмотрении графика растяжения кристалла по оси с (рис. 6б), выхода на плато функции изменения энергии ячейки ΔE не наблюдается. Таким образом, основное отличие в поведении объема V и изменения энергии ячейки ΔE для пластичного 3,4-дихлорфенола и эластичного 4-бром-3-хлорфенола состоят в том, что в случае пластичного кристалла его структура, начиная с определенного критического уровня растяжения, сравнительно долго остается мало изменяющейся, а эластичный кристалл продолжает оказывать сопротивление деформации, что вызывает осцилляции энергии, но при большом растяжении структура в нем все же перестраивается окончательно.
Следует отметить, что оба кристалла ведут себя практически одинаково при среднем и высоком растяжении, отличаясь только скоростью роста энергии ячейки и некоторыми особенностями нековалентных взаимодействий. Если сравнивать графики изменения энергии ячейки, то видно, что область с выходом на «плато» у пластичного кристалла меньше (см. рис. 3а), и в дальнейшем происходит рост ΔE, в то время, как у эластичного кристалла, во-первых, наблюдаются сравнительно длительные осцилляции ΔE, во-вторых, на начальной стадии выше скорость роста ΔE (см. рис. 6а). Это можно заметить, исходя из линейной аппроксимации начального участка (до 12 %) зависимости роста энергии ячейки от растяжения: коэффициент пропорциональности линейной зависимости ΔE от процента растяжения для 3,4-дихлорфенола ниже, чем для 4-бром-3-хлорфенола. Мы видим более сильное относительное увеличение энергии ячейки именно
а)
б)
Рис. 6. Зависимости объёма и изменения полной энергии кристаллографической ячейки относительно исходной оптимизированной структуры от процента удлинения каждого параметра ячейки 4-бром-3-хлорфенола: а – при растяжении вдоль осей a и b с шагом в 3 %; б – при растяжении вдоль оси с с шагом в 3 % для эластичного кристалла, что предполагает для него более вероятную релаксацию структуры до исходного состояния при снятии растягивающего напряжения. Более пологий тренд изменения энергии ячейки пластичного кристалла при растяжении, наоборот, означает вероятность сохранения структуры кристалла при снятии напряжения в некотором деформированном состоянии или её перехода в другую равновесную структуру, отличающуюся от начальной. Рассматривая же идентичное поведение этих кристаллов на протяжении большей части диапазона растяжения, мы можем утверждать о достижении эластичным кристаллом предела эластической деформации примерно на уровне 10 % растяжения. Предполагается, что при дальнейшем его растяжении вероятно его поведение уже как пластичной структуры.
Выводы
В результате применения теста на виртуальное растяжение, представляющего собой моделирование равновесных, но деформированных структур галогенфенолов, были даны объяснения пластичного и эластичного поведения этих кристаллических структур. В растянутой кристаллической структуре пластичного 3,4-дихлорфенола формируются сравнительно небольшие зазоры между стопками. Стопки, в свою очередь, с каждым новым шагом растяжения получают возможность разворачиваться друг относительно друга, сохраняя при этом синтоны с четырьмя водородными связями и «переключая» менее прочные межстопочные взаимодействия галогенов с типа I - ван-дер-ваальсова взаимодействия на более прочные - галогенные связи. С появлением зазоров между стопками изменение энергии кристаллической ячейки замедляется с выходом на короткое «плато», затем продолжает расти. Объем ячейки показывает монотонный рост во всем диапазоне растягивающих деформаций.
Изменение энергии и объема кристаллической ячейки при ее растяжении для эластичного кристалла 4-бром-3-хлорфенола происходит сходным образом, но лишь до определённого предела. Затем, с появлением значительных по размеру полостей между стопками в двух ортогональных направлениях, изменение энергии ячейки начинает осциллировать, а потом продолжает расти. Область замедления роста энергии в случае эластичного кристалла 4-бром-3-хлорфенола значительно больше, чем для пластичного 3,4-дихлорфенола, и она сопровождается осцилляциями. Это наблюдение можно интерпретировать как стремление эластичной структуры сохранять возможность возврата к предыдущему состоянию без разрушения.
Показано, что изменения энергии и объема ячейки кристаллов галогенфенолов при моделировании растягивающих деформаций мало зависит от режима растяжения - шага, изменяющегося в пределах от 1 до 3 %.
Таким образом, виртуальный тест на растяжение показал себя подходящим инструментом для объяснения механических свойств молекулярных кристаллов, в том числе, при сравнении эластичных и пластичных форм изоструктурных кристаллов, каковыми являются 3,4-дихлорфенол и 4-бром-3-хлорфенол.
Работа выполнена по гранту РФФИ №20-03-00240.
Список литературы Моделирование и анализ виртуального растяжения эластичного и пластичного кристаллов дигалогенфенолов
- Hohenberg P., Kohn W. Inhomogeneous Electron Gas. Phys. Rev. 1964;136:B864-B871. DOI: 10.1103/PhysRev.136.B864.
- Kohn W., Sham J.L. Self-Consistent Equations Including Exchange and Correlation Effects. Phys. Rev. 1965;140:A1133-A1138. DOI: 10.1103/PhysRev.140.A1133.
- Mattar V.T., Gavilan-Arriazu E.M., Rodriguez S.A. Study of Electrochemical Mechanisms Using Computational Simulations: Application to Phenol Butylated Hydroxyanisole. Journal of Chemical Education. 2022;99 (2):1044-1052. DOI: 10.1021/acs.jchemed.1c01230.
- Ngake T., Nqayi S., Gulumian M., Cronje S., Harris R.A. Recent developments in computational and experimental studies of physicochemical properties of Au and Ag nanostructures on cellular uptake and nanostructure toxicity. Biochimica et Biophysica Acta (BBA) - General Subjects. 2022;1866(8):130170. DOI: 10.1016/j.bbagen.2022.130170.
- Gupta P., Rather S.A., Saha B.K., Panda T. Mechanical Flexibility of Molecular Crystals Achieved by Exchanging Hydrogen Bonding Synthons. Cryst. Growth Des. 2020;20:2847-2852. DOI: 10.1021/acs.cgd.9b01530.
- Cook M.J., Wilson M.R. Calculation of Helical Twisting Power for Liquid Crystal Chiral Dopants. J. Chem. Phys. 2000;112 (3):1560-1564. DOI: 10.1063/1.480703.
- Earl D.J., Wilson M.R. Predictions of Molecular Chirality and Helical Twisting Powers: A Theoretical Study. J. Chem. Phys. 2003;119 (19):10280-10288. DOI: 10.1063/1.1617980.
- Wilson M.R., Earl D.J. Calculating the Helical Twisting Power of Chiral Dopants. J. Mater. Chem. 2001;11 (11):2672-2677. DOI: 10.1039/b103520g.
- Quevillon M. J., Whitmer J. K. Charge Transport and Phase Behavior of Imidazolium-based Ionic Liquid Crystals from Fully Atomistic Simulations. Materials (Basel). 2018;11 (1):64. DOI: 10.3390/ma11010064.
- Matveychuk Y.V, Bartashevich E.V., Skalyova K.K., Tsirelson V.G. Quantum electronic pressure and crystal compressibility for magnesium diboride under simulated compression. Materials Today Communications. 2021;26:101952. DOI: 10.1016/j.mtcomm.2020.101952.
- Bartashevich E.V., Sobalev S.A., Matveychuk Y.V., Tsirelson V.G. Simulation of the Compressibility of Isostructural Halogen Containing Crystals on Macro- and Microlevels. Journal of Structural Chemistry. 2021;62 (10):1607-1620. DOI: 10.1134/S0022476621100164.
- Bartashevich E.V., Sobalev S.A., Matveychuk Y.V., Tsirelson V.G. Variations of Quantum Electronic Pressure Under the External Compression in Crystals with Halogen Bonds Assembled in Cl3-, Br3-, I3-synthons. Acta Crystallographica Section B: Structural Science, Crystal Engineering and Materials. 2020;76:514-523. DOI: 10.1107/S2052520620006113.
- Masunov A.E., Wiratmo M., Dyakov A.A., Matveychuk Y.V. Virtual Tensile Test for Brittle, Plastic, and Elastic Polymorphs of 4-Bromophenyl 4-Bromobenzoate. Cryst. Growth Des. 2020;20:6093-6100. DOI: 10.1021/acs.cgd.0c00798.
- Desiraju G.R., Ho P.S., Kloo L., Legon A.C. Definition of the halogen bond. Pure Appl. Chem. 2013;85 (8): 1711-1713. DOI: 10.1351/PAC-REC-12-05-10.
- Metrangolo P., Resnati G. Halogen bonding: A paradigm in supramolecular chemistry. Chem. Eur. 2001;7:2511-2519. DOI: 10.1002/1521-3765(20010618)7:12<2511::AID-CHEM25110-3.0.CO;2-T.
- Metrangolo P., Neukirch H., Pilati T., Resnati G. Halogen bonding based recognition processes: A world parallel to hydrogen bonding. Acc. Chem. Res. 2005;38 (5): 386-395. DOI: 10.1021/ar0400995.
- Dovesi R., Erba A., Orlando R., Zicovich-Wilson C.M. Quantum-mechanical Condensed Matter Simulations with CRYSTAL. WIREs Comput. Mol. Sci. 2018;8: e1360. DOI: 10.1002/wcms.1360.
- Dovesi R. CRYSTAL17 User's Manual. University of Torino: Torino. 2017. 461 p.
- Cambridge Crystallographic Data Center. 2020. deposit@ccdc.cam.ac.uk; http://www.ccdc.cam.ac.uk
- Brandenburg J.G., Grimme S. Dispersion Corrected Hartree-Fock and Density Functional Theory for Organic Crystal Structure Prediction. Top. Curr. Chem. 2014;345:1-23. DOI: 10.1007/128_2013_488.
- Sure R., Grimme S. Corrected Small Basis Set Hartree-Fock Method for Large Systems. J. Comput. Chem. 2013;34:1672-1685. DOI: 10.1002/jcc.23317.
- Grimme S., Antony J., Ehrlich S., Krieg H. A Consistent and Accurate Ab Initio Parametriza-tion of Density Functional Dispersion Correction (DFT-D) for the 94 Elements H-Pu. J. Chem. Phys. 2010;132:154104. DOI: 10.1063/1.3382344.
- Kruse H., Grimme S. A Geometrical Correction for the Inter- and Intra-Molecular Basis Set Superposition Error in Hartree-Fock and Density Functional Theory Calculations for Large Systems. J. Chem. Phys. 2012;136:154101. DOI: 10.1063/1.3700154.
- Brandenburg J.G., Alessio M., Civalleri B., Peintinger M.F. Geometrical Correction for the Inter- and Intramolecular Basis Set Superposition Error in Periodic Density Functional Theory Calculations. J. Phys. Chem. A. 2013;117:9282-9292. DOI: 10.1021/jp406658y.
- Mukherjee A., Desiraju G.R. Halogen Bonds in Some Dihalogenated Phenols: Applications to Crystal Engineering. IUCrJ. 2013;1:49-60. DOI: 10.1107/S2052252513025657.
