Моделирование и изучение антикоронавирусной активности производных 6,7-диметоксихиназолин-4(3H)-она в отношении SARS-CoV-2 методами in silico и in vitro
-она в отношении SARS-CoV-2 методами in silico и in vitro")
Автор: Чиряпкин А.С., Тюренков И.Н., Кодониди И.П., Шабанова Н.Б., Вдовенко-мартынова Н.Н.
Журнал: Бюллетень науки и практики @bulletennauki
Рубрика: Медицинские науки
Статья в выпуске: 8 т.9, 2023 года.
Бесплатный доступ
Пандемия новой коронавирусной инфекции SARS-CoV-2 (COVID-19) оказала существенную нагрузку на систему здравоохранения и выявила необходимость разработки эффективных лекарственных препаратов для лечения данного заболевания. Приводимые в статье результаты исследования посвящены разработке новых соединений с антикоронавирусным действием в отношении SARS-CoV-2, которые являются производными 6,7-диметоксихиназолин-4(3H)-она. In silico исследования осуществляли методом молекулярного докинга в свободно распространяемой программе Autodock 4.0. Виртуальные структуры моделируемых соединений были построены в программе HyperChem 8.0.4, а окончательная оптимизация геометрии виртуальных структур рассчитывалась в программе ORCA 4.1 с использованием метода теории функционала плотности (UB3LYP) и базового набора 6-311G**. Для прогнозирования противовирусной активности использовалась главная протеаза (идентификационный номер 7K6D) и папаиноподобная (идентификационный номер 7LBR) протеаза SARS-Cov-2. In vitro исследования проводили путем кратных разведений анализируемых соединений с использованием хромогенного субстрата. Из результатов следует, что новые синтезированные производные 6,7-диметоксихиназолин-4(3H)-она, следует рассматривать в качестве перспективных кандидатов для разработки новых противовирусных лекарственных средств. Наиболее выраженная противовирусная активность наблюдается у вещества 8c, содержащего остаток глицилтриптофана. Стоит отметить, то предложенный алгоритм поиска инновационных противовирусных препаратов in silico достаточно хорошо согласуется с результатами эксперимента in vitro .
Sars-cov-2, ковид-19, 6, 7-диметоксихиназолин-4(3h)-оны, антикоронавирусная активность, молекулярный докинг, лиганд-ферментативный комплекс, протеазы
Короткий адрес: https://sciup.org/14128692
IDR: 14128692 | УДК: 615.281.8:519.876.5 | DOI: 10.33619/2414-2948/93/06
Modeling and study of anti-coronavirus activity of 6,7-dimethoxyquinazoline-4(3H)-one derivatives in relation to SARS-CoV-2 by in silico and in vitro methods
The pandemic of the new coronavirus infection SARS-CoV-2 (COVID-19) has put a significant strain on the healthcare system and revealed the need to develop effective medicines for the treatment of this disease. The research results presented in the article are devoted to the development of new compounds with anti-coronavirus action against SARS-CoV-2, which are derivatives of 6,7-dimethoxykinazoline-4(3H)-one. In silico, the research was carried out by the molecular docking method in the freely distributed Autodock 4.0 program. The virtual structures of the simulated compounds were constructed in the HyperChem 8.0.4 program, and the final optimization of the geometry of virtual structures was calculated in the ORCA 4.1 program using the density functional theory (UB3LYP) method and the base set 6-311G**. The main protease (identification number 7K6D) and papain-like (identification number 7LBR) protease SARS-Cov-2 were used to predict antiviral activity. In vitro studies were carried out by multiple dilutions of the analyzed compounds using a chromogenic substrate. It follows from the results that the newly synthesized derivatives of 6,7-dimethoxyquinazoline-4(3H)-one should be considered as promising candidates for the development of new antiviral drugs. The most pronounced antiviral activity is observed in substance 8c containing a glycyltryptophan residue. It is worth noting that the proposed algorithm for searching for innovative antiviral drugs in silico agrees quite well with the results of the in vitro experiment.
Текст научной статьи Моделирование и изучение антикоронавирусной активности производных 6,7-диметоксихиназолин-4(3H)-она в отношении SARS-CoV-2 методами in silico и in vitro
Бюллетень науки и практики / Bulletin of Science and Practice
Появление новой коронавирусной инфекции (COVID-19/SARS-CoV-2) в декабре 2019 года потребовало реализацию задач по поиску и созданию новых противовирусных препаратов. Глобальное распространение SARS-CoV-2 в совокупности с высоким количеством смертей побудило Всемирную организацию здравоохранения объявить пандемию 12 марта 2020 года [1]. За время пандемии погибло около 20 миллионов человек. В настоящее время на фармацевтическом рынке отсутствуют высокоэффективные лекарственные средств против данного инфекционного заболевания, соответственно это подчеркивает актуальность проблемы по разработки высокоэффективных и безопасных веществ против инфекции SARS-CoV-2 [2]. Значимыми мишенями в поиске соединений для терапии SARS-CoV-2 являются главная и папаино-подобная протеазы COVID-19, ингибирование которых приводит к нарушению жизнедеятельности вируса и его гибели [3].
Конденсированные и бензаннелированные производные пиримидина широко используются в медицинской практике для лечения различных заболеваний. В частности пиримидиновое ядро содержится в структурах различных противовирусных лекарственных препаратов [4], что объясняет целесообразность поиска новых противовирусных биологически активных соединений среди производных пиримидина. Например, синтезирована серия производных индоло[3,2-c]изохинолина (δ-карболина), некоторые из которых обладают противомикробным действием, а дальнейшее изучение этих структур in silico позволило предположить возможность ими ингибировать главную протеазу вируса COVID-19 [5]. In vitro установлено, что новые аналоги пиримидиновых тиогликозидов обладают активностью против штаммов вируса SARS-COV-2 и птичьего гриппа H5N1 [6]. Пиридо[2,3-d]пиримидины могут более эффективно ингибировать главную протеазу SARS-COV-2, чем препарат лопинавир, который обычно используется в качестве ингибитора вирусных протеаз [7]. Новые гетероциклические конденсированные системы содержащие фрагменты пиримидина и бензимидазола можно рассматривать в качестве ингибиров главной протеазы и спайкового гликопротеина SARS-CoV-2 [8]. Согласно данным in silico некоторые представители синтезированных нафтопирано[2,3-d]пиримидин-11(12H)-онов, могут обладать высоким сродством к сайту связывания главной протеазы коронавируса, что обуславливает их фармакологическое действие [9]. Таким образом, конденсированные гетероциклические производные пиримидина являются перспективным классом органических соединений для поиска новых биологически активных соединений с антикоронавирусной активностью. Среди методов прогнозирования биологической активности соединений in silico выделяют наиболее значимый и широко используемый метод молекулярного докинга. В ходе такого вычислительного эксперимента представляется возможным предположить энергии взаимодействия виртуальных структур с биологической мишенью и следовательно более обоснованно построить алгоритм исследования соединений in vitro и in vivo [10].
Материал и методы исследования
Объектами виртуального исследования выступают производные 6,7-диметоксихиназолин-4(3H)-она с остатками нейроактивных аминокислот и дипептидов [11]. Структурные формулы изучаемых соединений приведены на Рисунке 1 под шифрами 1с-13с.
Молекулярный докинг проводился в свободно распространяемой программе Autodock 4.0 [12]. Молекулярное моделирование осуществлялось с учетом конформационной подвижности лигандов, торсионные углы которых были установлены и заданы в данной программе. Заряды всех атомов моделируемой системы рассчитаны алгоритмом гастейгера (Gasteiger algorithm). В программе был задан поиск 200 энергетически выгодных конформаций образования молекулярного комплекса изучаемых соединений с белковой мишенью с использованием ламарковской скоринговой функцией подсчета энергии взаимодействия (Lamarckian GA 4.2). Расстояние между точками сетки составляло 0,375 ангстрема. Трехмерные модели рецепторов и ферментов для вычислительных экспериментов брали из базы данных виртуальных структур RCSB Protein Data Bank ( [13].
Виртуальные структуры моделируемых соединений были построены в программе HyperChem 8.0.4, а затем геометрически оптимизированы способом молекулярной механики с использованием метода MM+ [14]. Окончательная оптимизация геометрии виртуальных структур рассчитывалась в программе ORCA 4.1 с использованием теории функционала плотности (UB3LYP) и базового набора 6-311G**.
Преобразование формата hin в pdb, необходимого для молекулярного моделирования, было выполнено в программе Open Babel 2.4.1 [15].
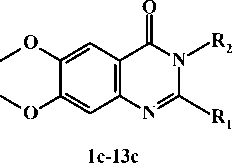
1с; R = -C H , R = -CH -COOH
2c; R = -C H , R =-CH -C(O)-NH-CH -COOH
5c; R = -C H , R = -CH -COOH
6c; R = -C H , R = -CH -C(O)-NH-CH -COOH
3c; R1 = -C2H5, R
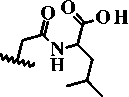
7c; R1 = -C3H7, R2
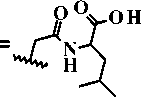
4c; R1 = -C2H5, R2 =
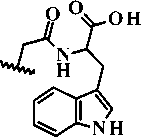
8c; R1 = -C3H7, R
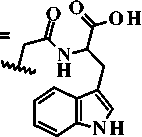
9c; R1 = -CH(CH3)2, R2 = -CH2-C(O)-NH-CH2-COOH
11c; R = -CH ; R = -CH COOH
12 с ; R = Ph; R = -CH COOH
13 с ; R = Ph; R = -C H COOH
10c; R1 = -CH(CH3)2, R2 =
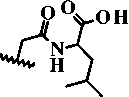
Рисунок 1. Структурные формулы прогнозируемых и далее синтезированных производных 6,7-диметоксихиназолин-4(3H)-она
Для прогнозирования противовирусной активности использовалась главная протеаза (идентификационный номер 7K6D [16]) и папаино-подобная (идентификационный номер 7LBR [17]) протеаза SARS-Cov-2. Областью вычислительного эксперимента для главной протеазы является куб, центр которого находится по следующим координатам: x = 9,664, y = 1,911, z = 22,528. Количество точек в измерениях по осям x, y и z равно 46. В случае папаиноподобной протеазы областью моделирования также является куб, центр которого находится по следующим координатам: x = 2,285, y = 14,435, z = 34,815. Количество точек в измерениях по осям x и z равно 46, а по оси y составляет 52.
Целенаправленный синтез наиболее перспективных прогнозируемых соединений выявленных in silico раннее был подробно описан в статье [11]. Соединения синтезировали взаимодействием соответствующих бензоксазинонов с глицином, глицилглицином, глициллейцином и глицилтриптофаном в среде ледяной уксусной кислоты при добавлении диметилформамида.
Оценку ингибирующей активности в отношении главной (Mpro) и папаино-подобной протеазы (PLpro) SARS-Cov-2 определяли путем проведения in vitro тестов [18, 19]. Для этого 20 мг исследуемого соединения растворяли в ДМСО до получения маточного раствора с концентрацией 1 мг/мл. Далее приготовляли серию двукратных разведений (растворитель — ДМСО) с конечным содержанием исследуемого соединения 500 µг/мл; 250 µг/мл; 125 µг/мл; 62,5 µг/мл; 31,25 µг/мл; 15,625 µг/мл и 7,8125 µг/мл. В микропланшет вносили 20 µМ Mpro или PLpro SARS-Cov-2 (Sigma-Aldrich) в объеме 10 µл, добавляли 10 µл раствора двукратных разведений исследуемого соединения. Полученную смесь инкубировали 10 минут при температуре 37 °С после чего в анализируемую среду вносили хромогенный субстрат и доводили до 200 µл буферным раствором 20 мМ HEPES рН 6,0 + 0,4 мМ ЭДТА+ 1 мМ дитиотреитол + 1% глицерин. Полученную смесь повторно инкубировали 60 минут в аналогичном температурном режиме. Параллельно ставили контрольную пробу и холостую пробу с водой для инъекций. Оптическую плотность полученных смесей регистрировали при 450 нм с использованием системы микропланшетного ридера Infinite F50 (Tecan, Австрия). Степень ингибирования ферментативной активности рассчитывали по разнице оптической плотности опытной/контрольной и холостой проб в %. На основании полученных результатов строили кривую зависимости «ингибирующая активность — logC» и рассчитывали коэффициент полуингибиции (IC50) в µМ/мл методом пробит-анализа. Все тесты выполнены в триплетном варианте с расчетом M±SEM (среднее ± стандартная ошибка среднего)
Результаты и обсуждение
С целью выявления достоверности вычислительного эксперимента связывания виртуальных структур с белковой мишенью осуществлялся расчет лиганд-ферментных комплексов классических ингибиторов протеаз и сравнение его с экспериментально полученными результатами рентгеноструктурного анализа описанными в научной литературе [16, 17]. В результате сравнительного анализа конформаций экспериментально определенных и теоретически рассчитанных лиганд-ферментных комплексов известных ингибиторов установлена достаточная достоверность используемого метода моделирования. Такой вывод вытекает из сравнения пространственное расположение референта телапревира в эксперименте in silico и экспериментально полученного рентгеноструктурным анализом, эта разница равняется 1,84 ангстремом (Рисунок 2), а для референтного вещества XR8-89 равняется 1,81 ангстрем (Рисунок 3). Эта разница меньше классически описанного предельного порога 3,0 ангстрем. Тем самым полученные результаты in silico эксперимента обладают достаточно высокой достоверностью.
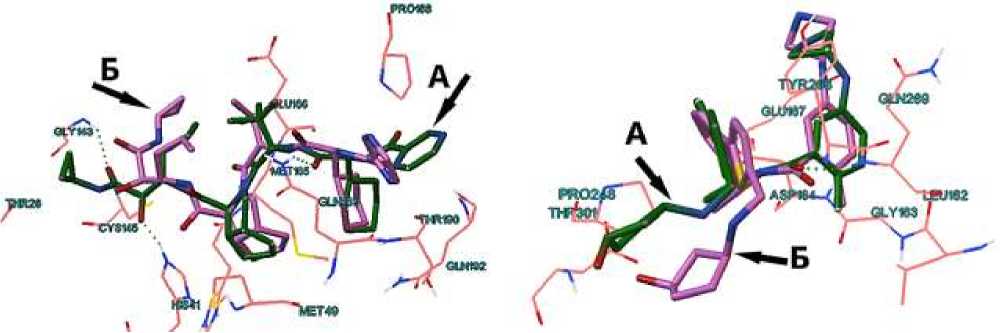
Рисунок 2. Расположение телапревира в активном сайте главной протезы SARS-Cov-2. А — рентгеноструктурный анализ (зеленый цвет), Б — молекулярный докинг (розовый цвет)
Рисунок 3. Расположение XR8-89 в активном сайте папаино-подобной протезы SARS-Cov-2. А — рентгеноструктурный анализ (зеленый цвет), Б — молекулярный докинг (розовый цвет)
Наибольшее сродство к активному сайту главной протеазы SARS-Cov-2 показали производные 6,7-диметоксихиназолин-4(3H)-она с остатками глициллейцина и глицилтриптофан: 3c, 4c, 7c и 8c (Таблица 1). Максимальной устойчивостью лиганд-ферментного комплекса обладает вещество 2-пропил-6,7-диметокси-3,1-бензоксазин-4-она содержащее фрагмент глицилтриптофана (-9,38 ккал/моль, 8с). Особо следует отметить, что для наиболее перспективных структур (3с, 7с и 8с) представляется возможным формирование межмолекулярной водородной связи с Glu 166. На Рисунке 4 приведена наиболее устойчивая конформация соединения-лидера 8с в активном сайте главной протеазой SARS-Cov-2.
Таблица 1
ЗНАЧЕНИЯ ЭНЕРГИЙ ЛИГАНД-ФЕРМЕНТНОГО КОМПЛЕКСА
И ДЛИН ВОДОРОДНЫХ СВЯЗЕЙ ПРОИЗВОДНЫХ
6,7-ДИМЕТОКСИХИНАЗОЛИН-4(3 H )-ОНА С ГЛАВНОЙ ПРОТЕАЗОЙ SARS-COV-2
|
Соединение |
Энергия докинга, ккал/моль |
Водородные связи |
||
|
Остаток аминокислоты |
Группа лиганда |
Длина водородной связи, Å |
||
|
1c |
-6,24 |
Gln192 |
COOH |
1,731 |
|
2c |
-7,75 |
Glu 166 |
COOH |
2,055 |
|
Gln 189 |
C=O |
2,197 |
||
|
3c |
-8,21 |
Glu 166 |
COOH |
2,198 |
|
4c |
-9,05 |
Gln 189 |
C=O |
1,901 |
|
Gln 192 |
COOH |
2,095 |
||
|
5c |
-6,20 |
Gln 189 |
C=O |
2,124 |
|
6c |
-7,22 |
— |
— |
— |
|
7c |
-8,59 |
Glu 166 |
COOH |
2,225 |
|
8c |
-9,38 |
Gly 143 |
OCH 3 |
2,189 |
|
Glu 166 |
C=O |
1,759 |
||
|
9c |
-7,22 |
Gln 189 |
COOH |
2,016 |
|
10c |
-7,89 |
Gln 189 |
C=O |
2,073 |
|
Glu 166 |
COOH |
1,173 |
||
|
11c |
-6,13 |
Gln 192 |
COOH |
2,227 |
|
12c |
-6,81 |
Arg 188 |
C=O |
2,185 |
|
13c |
-7,03 |
Gly 143 |
COOH |
2,252 |
|
Glu 166 |
-N= |
1,926 |
||
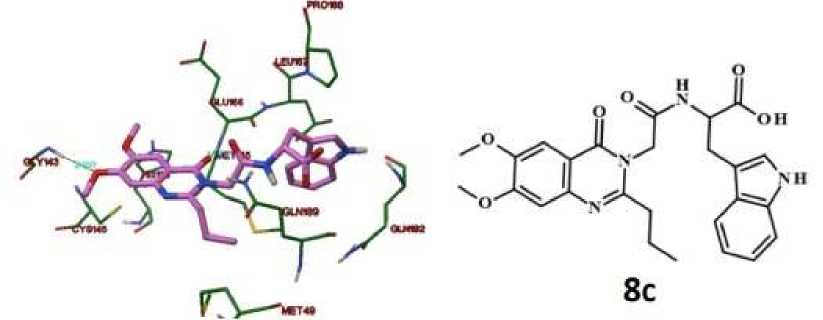
Рисунок 4. Расположение соединения 8с в активном сайте главной протеазы SARS-Cov-2
Как и в случае с главной протеазой наибольшее сродство к папаино-подобной протеазе SARS-Cov-2 прогнозируется для производных 6,7-диметоксихиназолин-4(3 H )-она с остатками глициллейцина и глицилтриптофана, вещества 4с, 7с и 8с (Таблица 2).
Таблица 2
ЗНАЧЕНИЯ ЭНЕРГИЙ ЛИГАНД-ФЕРМЕНТНОГО КОМПЛЕКСА
И ДЛИН ВОДОРОДНЫХ СВЯЗЕЙ ПРОИЗВОДНЫХ 6,7-ДИМЕТОКСИХИНАЗОЛИН-4(3 H )-ОНА С ПАПАИНО-ПОДОБНОЙ ПРОТЕАЗОЙ SARS-COV-2
|
Соединение |
Энергия докинга, ккал/моль |
Водородные связи |
||
|
Остаток аминокислоты |
Группа лиганда |
Длина водородной связи, Å |
||
|
1с |
-5,22 |
Arg 166 |
-COOH |
1,981 |
|
2с |
-6,55 |
Asn 267 |
-COOH |
2,135 |
|
Tyr 268 |
-COOH |
1,916 |
||
|
3с |
-6,75 |
Tyr 268 |
-COOH |
1,910 |
|
4с |
-7,56 |
Trp 1 |
-СОNH- |
1,984 |
|
5с |
-5,67 |
Thr 301 |
-COOH |
2,126 |
|
Arg 166 |
-COOH |
2,169 |
||
|
6с |
-6,75 |
Tyr 268 |
-COOH |
2,013 |
|
7с |
-7,29 |
Gly 266 |
-COOH |
2,053 |
|
8с |
-8,82 |
— |
— |
— |
|
9с |
-6,52 |
— |
— |
— |
|
10с |
-6,86 |
Gln 269 |
-COOH |
1,803 |
|
11с |
-5,30 |
Tyr 268 |
-COOH |
1,859 |
|
12с |
-7,15 |
Thr 301 |
-COOH |
2,007 |
|
13с |
-8,11 |
Arg 166 |
-COOH |
1,856 |
|
Arg 166 |
COOH |
2,041 |
||
|
Телапривир |
-7,52 |
Arg 166 |
-N= |
2,113 |
Однако структура 13с, содержащая остаток β-аланина, предположительно обладает соизмеримой энергией взаимодействия в сравнении с соединениями-лидерами (-8,11 ккал/моль, 13с). Экспериментальные исследования in vitro производные 6,7-диметоксихиназолин-4(3H)-она показали достаточно высокую способность ингибировать протеазы SARS-Cov-2 (Таблица 3). Наиболее выраженная активность наблюдается у вещества 8с, содержащего остаток глицилтриптофана. Причем согласно литературным данным, соединения-лидеры превосходят по активности известный препарат эбселен, для которого концентрация полумаксимального ингибирования для главной протезы приводится равная 0,67±0,09 мкМ/мл, а для папаиноподобной — 0,52±0,06 мкМ/мл [20].
ВЛИЯНИЕ ИССЛЕДУЕМЫХ СОЕДИНЕНИЙ
НА ИНГИБИРОВАНИЕ MPRO И PLPRO SARS-COV-2
Таблица 3
|
Соединение |
IC50, мкМ/мл |
Соединение |
IC50, мкМ/мл |
|
Ингибирование главной протеазы SARS-Cov-2 |
|||
|
1c |
0,09±0,008 |
8c |
0,012±0,0048 |
|
2c |
0,09±0,008 |
9c |
0,098±0,0076 |
|
3c |
0,1±0,007 |
10c |
0,062±0,0004 |
|
4c |
0,08±0,009 |
11c |
0,046±0,001 |
|
5c |
0,062±0,0015 |
12c |
0,09±0,004 |
|
6c |
0,081±0,0046 |
13c |
0,1±0,007 |
|
7c |
0,032±0,0087 |
эбселен |
0,67±0,09 [20] |
Бюллетень науки и практики / Bulletin of Science and Practice Т. 9. №8. 2023
|
Соединение |
IC50, мкМ/мл |
Соединение |
IC50, мкМ/мл |
|
Ингибирование папаино |
подобной протеазы SARS-Cov-2 |
||
|
1c |
0,08±0,002 |
8c |
0,013±0,0093 |
|
2c |
0,07±0,004 |
9c |
0,041±0,001 |
|
3c |
0,07±0,004 |
10c |
0,074±0,0038 |
|
4c |
0,05±0,006 |
11c |
0,023±0,002 |
|
5c |
0,016±0,0006 |
12c |
0,07±0,007 |
|
6c |
0,025±0,0072 |
13c |
0,1±0,003 |
|
7c |
0,018±0,001 |
эбселен |
0,52±0,06 [20] |
В результате проведенного исследования осуществлено молекулярное конструирование и дальнейший целенаправленный синтез производных 6,7-диметоксихиназолин-4(3H)-она с остатками нейроактивных аминокислот и дипептидов обладающих антикоронавирусным действием. Полученные вещества следует рассматривать в качестве перспективных кандидатов для разработки новых противовирусных лекарственных средств. Предложенный алгоритм поиска инновационных противовирусных препаратов in silico достаточно хорошо согласуется с результатами эксперимента in vitro .
Финансирование: Исследование выполнено при финансовой поддержке РФФИ в рамках научного проекта №20-315-90060.
Financing . The reported study was funded by RFBR, project no. 20-315-90060.
Список литературы Моделирование и изучение антикоронавирусной активности производных 6,7-диметоксихиназолин-4(3H)-она в отношении SARS-CoV-2 методами in silico и in vitro
- Wu D., Wu T., Liu Q., Yang Z. The SARS-CoV-2 outbreak: what we know // International journal of infectious diseases. 2020. V. 94. P. 44-48. https://doi.org/10.1016/j.ijid.2020.03.004
- Pozzi C., Vanet A., Francesconi V., Tagliazucchi L., Tassone G., Venturelli A., Tonelli M. Antitarget, Anti-SARS-CoV-2 Leads, Drugs, and the Drug Discovery–Genetics Alliance Perspective // Journal of Medicinal Chemistry. 2023. V. 66. №6. P. 3664-3702. https://doi.org/10.1021/acs.jmedchem.2c01229
- Золотых Д. С., Дайронас Ж. В., Поздняков Д. И. Противовирусная активность соединений, содержащих структурный фрагмент бензопиран-2-она // Вопросы биологической, медицинской и фармацевтической химии. 2022. Т. 25. №6. С. 22-27. https://doi.org/10.29296/25877313-2022-06-03
- Чиряпкин А. С. Обзор производных пиримидина как фармакологически активных соединений // Juvenis Scientia. 2022. Т. 8. №5. С. 16-30. https://doi.org/10.32415/jscientia_2022_8_5_16-30
- Verma V. A., Saundane A. R., Meti R. S., Vennapu D. R. Synthesis of novel indolo [3, 2-c] isoquinoline derivatives bearing pyrimidine, piperazine rings and their biological evaluation and docking studies against COVID-19 virus main protease // Journal of Molecular Structure. 2021. V. 1229. P. 129829. https://doi.org/10.1016/j.molstruc.2020.129829
- Abu-Zaied M. A., Elgemeie G. H., Mahmoud N. M. Anti-covid-19 drug analogues: synthesis of novel pyrimidine thioglycosides as antiviral agents against SARS-COV-2 and avian influenza H5N1 viruses // ACS omega. 2021. Vol. 6. №26. P. 16890-16904. https://doi.org/10.1021/acsomega.1c01501
- Alamshany Z. M., Khattab R. R., Hassan N. A., El-Sayed A. A., Tantawy M. A., Mostafa A., Hassan A.A. Synthesis and Molecular Docking Study of Novel Pyrimidine Derivatives against COVID-19 // Molecules. 2023. Vol. 28. №2. P. 739. https://doi.org/10.3390/molecules28020739
- Khan S., Kale M., Siddiqui F., Nema N. Novel pyrimidine-benzimidazole hybrids with antibacterial and antifungal properties and potential inhibition of SARS-CoV-2 main protease and spike glycoprotein // Digital Chinese Medicine. 2021. V. 4. №2. P. 102-119. https://doi.org/10.1016/j.dcmed.2021.06.004
- Algethami F. K., Cherif M., Jlizi S., Ben Hamadi N., Romdhane A., Elamin M. R., ... & Ben Jannet H. Design, Microwave-Assisted Synthesis and In Silico Prediction Study of Novel Isoxazole Linked Pyranopyrimidinone Conjugates as New Targets for Searching Potential Anti- SARS-CoV-2 Agents // Molecules. 2021. V. 26. №20. P. 6103. https://doi.org/10.3390/molecules26206103
- Кодониди И. П., Чиряпкин А. С., Творовский Д. Е. Молекулярное конструирование N-ацильных производных 2-(2-оксопиролидин-1-ил)-ацетамида, обладающих ГАМК-ергической и глутаматергической активностями // Фармация и фармакология. 2021. Т. 9. №1. С. 84-97. https://doi.org/10.19163/2307-9266-2021-9-1-84-97
- Chiriapkin A. S., Kodonidi I. P., Pozdnyakov D. I. Synthesis and evaluation of cerebroprotective activity of novel 6,7-dimethoxyquinazolin-4(3H)-one derivatives containing residues of amino acids and dipeptides // Chimica Techno Acta. 2022. Vol. 9. №2. P. 20229212. https://doi.org/10.15826/chimtech.2022.9.2.12
- Morris G. M., Huey R., Lindstrom W., Sanner M. F., Belew R. K., Goodsell D. S., Olson A. J. Autodock4 and AutodockTools4: Automated Docking with Selective Receptor Flexibility // J. Comput. Chem. 2009. V. 30. P. 2785-2791. https://doi.org/10.1002/jcc.21256
- Berman H. M., Westbrook J., Feng Z., Gilliland G., Bhat T. N., Weissig H., Bourne P. E. The Protein Data Bank // Nucleic Acids Res. 2000. V. 28. №1. P. 235-242. https://doi.org/10.1093/nar/28.1.235
- Brian J. T. Hyperchem, release 2: molecular modeling for the personal computer // J. Chem. Inf. Comput. Sci. 1992. V. 32. P. 757–759. https://doi.org/10.1021/ci00010a025
- O'Boyle N. M. Banck M., James C. A., Morley C., Vandermeersch T., Hutchison G. R. Open Babel: An open chemical toolbox // J. Cheminform. 2011. V. 3. №1. P. 33. https://doi.org/10.1186/1758-2946-3-33
- Andi B., Kumaran D., Kreitler D. F., Soares A. S., Keereetaweep J., Jakoncic J., .McSweeney S. Hepatitis C virus NS3/4A inhibitors and other drug-like compounds as covalent binders of SARS-CoV-2 main protease // Sci. Rep. 2022. V. 12. P. 12197. https://doi.org/10.1038/s41598-022-15930-z
- Shen Z., Ratia K., Cooper L., Kong D., Lee H., Kwon Y., Xiong R. Design of SARSCoV- 2 PLpro Inhibitors for COVID-19 Antiviral Therapy Leveraging Binding Cooperativity // J. Med. Chem. 2022. V. 65. №4. P. 2940-2955. https://doi.org/10.1021/acs.jmedchem.1c01307
- Yang H., Xie W., Xue X., Yang K., Ma J., Liang W., Rao Z. Design of wide-spectrum inhibitors targeting coronavirus main proteases // PLoS Biol. 2005. V. 3. №11. P. e428. https://doi.org/10.1371/journal.pbio.0030428
- Pitsillou E., Liang J., Ververis K., Lim K. W., Hung A., Karagiannis T. C. Identification of small molecule inhibitors of the deubiquitinating activity of the SARS-CoV-2 papain-like protease: in silico molecular docking studies and in vitro enzymatic activity assay // Front. Chem. 2020. V. 8. P. 623971. https://doi.org/10.3389/fchem.2020.623971
- Amporndanai K., Meng X., Shang W., Jin Z., Rogers M., Zhao Y., Samar Hasnain S. Inhibition mechanism of SARS-CoV-2 main protease by ebselen and its derivatives // Nat. Commun. 2021. V. 12. №1. P. 3061. https://doi.org/10.1038/s41467-021-23313-7
