Молекулярно-генетические механизмы сигнального каскада RAS-RAF-MEK-ERK, связанные с развитием опухолевого процесса и назначением таргетных препаратов при колоректальном раке

Автор: Тороповский А.Н., Павлова О.Н., Викторов Д.А., Никитин А.Г.
Журнал: Вестник медицинского института "РЕАВИЗ": реабилитация, врач и здоровье @vestnik-reaviz
Рубрика: Морфология. Патология
Статья в выпуске: 4 (52), 2021 года.
Бесплатный доступ
Колоректальный рак занимает одну из лидирующих позиций в мире в структуре онкологической заболеваемости. Процессы жизнедеятельности раковых клеток во многом зависят от продукции ростовых факторов и их рецепторов. Одним из таковых является эпидермальный фактор роста (EGFR), представляющий собой тирозин-киназный рецептор мембран клеток. В норме связывание лигандов EGFR и трансформирующего фактора роста альфа (TGFα) индуцирует активацию рецепторов, что запускает ERK и PI3K сигнальные пути, контролирующие клеточную пролиферацию, миграцию, инвазию и множество других процессов. Установлено, что в 80 % случаев колоректальный рак возникает в результате гиперэкспрессии EGFR, которая приводит к усиленному росту и делению клеток опухоли вследствие гиперактивации RAS-RAF-MEK-ERK сигнального каскада. Каскад RAS-RAF-MEK-ERK является путем, который регулирует клеточную пролиферацию, клеточный цикл и миграцию клетки. При развитии рака у человека мутации семейства RAS/RAF наиболее часто являются причиной нарушения регуляции трансдукции сигнала через этот путь. Согласно современным данным около трети всех злокачественных новообразований ассоциированы с мутациями в генах семейства RAS, которые включают в себя HRAS, KRAS, NRAS, RRAS и другие гомологичные белки. Белки семейства RAS принимают участие в активации сигнальных путей тирозин-киназы, что приводит к мутации генов. Этот процесс определяет пролиферативную активность, способность к дифференцировке, метастазирование, уход от апоптоза, индукцию ангиогенеза. Постоянная активация RAS ведет к злокачественному перерождению клеток. Таким образом, экспрессия и мутация гена EGFR связаны с различными вариантами прогрессирования опухоли и неблагоприятным прогнозом при злокачественных новообразованиях различных локализаций. За последние десятилетия достигнуты значительные успехи в лечении метастатического колоректального рака. Однако расширение спектра эффективных противоопухолевых препаратов формирует и ряд сложностей при выборе оптимальных схем лекарственной терапии у пациентов с метастазами колоректального рака.
Колоректальный рак, рак толстой и прямой кишки, метастазированный колоректальный рак, egfr, h-ras, kras, nras, r-ras, ras-raf-mek-erk сигнальный каскад
Короткий адрес: https://sciup.org/143177460
IDR: 143177460 | УДК: 616-006.446.8 | DOI: 10.20340/vmi-rvz.2021.4.MORPH.3
Molecular-genetic mechanisms of the signal cascade RAS-RAF-MEK-ERK associated with the development of the tumor process and the purpose of targeted drugs for colorectal cancer
Colorectal cancer occupies one of the leading positions in the world in the structure of cancer incidence. The vital processes of cancer cells largely depend on the production of growth factors and their receptors. One of these is epidermal growth factor (EGFR), which is a tyrosine kinase receptor for cell membranes. Normally, binding of EGFR ligands and transforming growth factor alpha (TGFα) induces receptor activation, which triggers ERK and PI3K signaling pathways that control cell proliferation, migration, invasion, and many other processes. It was found that in 80% of cases, colorectal cancer occurs as a result of EGFR overexpression, which leads to increased growth and division of tumor cells due to hyperactivation of the RAS-RAF-MEKERK signaling cascade. The RAS-RAF-MEK-ERK cascade is a pathway that regulates cell proliferation, cell cycle, and cell migration. In the development of human cancer, mutations of the RAS/RAF family are most often the cause of dysregulation of signal transduction through this pathway. According to current data, about a third of all malignant neoplasms are associated with mutations in the genes of the RAS family, which include HRAS, KRAS, NRAS, RRAS, and other homologous proteins. Proteins of the RAS family are involved in the activation of tyrosine kinase signaling pathways, which leads to gene mutations. This process determines proliferative activity, ability to differentiate, metastasis, avoidance of apoptosis, induction of angiogenesis. Permanent RAS activation leads to malignant cell degeneration. Thus, the expression and mutation of the EGFR gene are associated with various variants of tumor progression and poor prognosis in malignant neoplasms of various localizations. Over the past decades, significant advances have been made in the treatment of metastatic colorectal cancer. However, the expansion of the spectrum of effective anticancer drugs also creates a number of difficulties in choosing the optimal drug therapy regimens in patients with metastases of colorectal cancer.
Текст научной статьи Молекулярно-генетические механизмы сигнального каскада RAS-RAF-MEK-ERK, связанные с развитием опухолевого процесса и назначением таргетных препаратов при колоректальном раке
Cite as: Toropovsky A.N., Pavlova O.N., Viktorov D.A., Nikitin A.G. Molecular-genetic mechanisms of the signal cascade RAS-RAF-MEK-ERK associated with the development of the tumor process and the purpose of targeted drugs for colorectal cancer. Bulletin of the Medical Institute REAVIZ. Rehabilitation, Doctor and Health. 2021;4(52):25-35.
Рак толстой и прямой кишки (РТПК) или колоректальный рак (КРР) является одним из самых распространенных злокачественных новообразований у человека. В России отмечается неуклонный рост заболеваемости и смертности от КРР. По данным В.П. Земляного ежегодно в мире регистрируется около 800 000 пациентов с колоректальным раком и 440 000 смертей от этого заболевания [1]. По статистическим данным на 2015 год в России диагностировали КРР у более чем 68 000 человек, а смертность составила – более 42 000 случаев [2]. Таким образом, КРР занимает 3-е место по заболеваемости как у мужчин, так и у женщин и 3-е место в структуре смертности от онкологических заболеваний у мужчин и 2-е – у женщин [3]. Причем среди вариантов колоректального рака наиболее встречаемым и смертоносным является рак толстой кишки (РТК), так как это заболевание может значительное время протекать бессимптомно и, как следствие, у каждого третьего больного на момент установления диагноза отмечается генерализация опухолевого процесса. Высокая смертность при РТК объ- ясняется частым метастазированием опухоли, в основном в печень и легкие, нередко даже на первых стадиях заболевания [4].
Основой многих онкологических заболеваний являются нарушения в регуляции клеточного цикла, которые приводят к безудержному росту клеток и формированию опухолей. Причины нарушений клеточного деления заключаются чаще всего в соматических мутациях – изменениях исходной генетической информации соматических клеток. Нарушение экспрессии генов и различные хромосомные перестройки (транслокации, делеции, инверсии и амплификации) могут возникнуть в структуре протоонкогенов, генах ростовых факторов, клеточных рецепторов и других биологически активных генах. Данные нарушения приводят к потере контроля над работой протоонкогенов в составе нормального генома и запускают процессы злокачественной трансформации. Нарушения экспрессии белков, контролирующих процессы апоптоза, пролиферации и ангиогенеза, эпигенетические изменения ДНК белков, контролирующих жизнедеятельность опухолевых клеток, влияют на клиническое течение опухолевого процесса. Доказано, что различия в экспрессии определенных белковых маркеров могут объяснить, почему сравнимые по стадии, гистологической структуре и степени злокачественности опухоли различаются по течению заболевания. Раннюю диагностику рака толстой кишки до настоящего времени нельзя считать удовлетворительной, так как выявляе-мость опухоли на ранних стадиях низкая [5].
Проведенный анализ литературы показывает, что пусковым механизмом онкогенеза большинства новообразований толстой кишки являются нарушения генетического аппарата клетки. В то же время существует значительный разрыв между научно-обоснованными данными в области молекулярной генетики канцерогенеза и практическим применением их в работе врачей [6].
Рак толстой кишки (РТК) представляет собой гетерогенную группу опухолей, отличающихся как механизмами канцерогенеза и, следовательно, молекулярными изменениями, так и прогнозом течения болезни, и особенностями терапии. Уже сейчас для выбора тактики лечения необходимо учитывать не только клинические факторы, такие как распространение опухоли, функциональный статус пациента, но и молекулярный профиль заболевания.
Одна из основных причин, влияющих на развитие этого заболевания, – эпигенетические нарушения регуляции генов. Определение этих нарушений имеет большое значение для ранней диагностики раковых заболеваний, так как нарушенная регуляция генов вследствие аберрантного метилирования ДНК является ключевой стадией в развитии опухоли. При этом, метилированная ДНК является достаточно информативным биомаркером и может быть легко измерена в образцах крови или плазмы. Метилирование обширных регионов CpG-островков – основное эпигенетическое изменение, характерное для процессов канцерогенеза при КРР [7].
Важно отметить, что маркерные молекулы для диагностики заболеваний не всегда лежат в основе развития заболеваний. Данное утверждение в особенности справедливо для маркеров метилирования. В ряде работ было показано отсутствие прямой корреляции между аберрантным метилированием и выраженностью экспрессии генов [8, 9].
В патогенезе КРР главным является пролиферация эпителиальных клеток слизистой оболочки кишечника. Ранее было показано, что в 80 % случаев РТПК есть гиперэкспрессия EGFR, которая приводит к усиленному росту и делению клеток опухоли вследствие гиперактивации RAS-RAF-MEK-ERK сигнального каскада [10].
RAS-RAF-MEK-ERK сигнальный путь представляет собой цепь последовательно взаимодействующих белков, которые передают сигнал с поверхности клетки от кле- точного рецептора внутрь ядра клетки к ДНК. Этот сигнальный путь включает в себя много белков, в том числе MEK (Mitogen-activated protein kinase). Участники сигнального пути взаимодействуют между собой с помощью фосфорилировния и дефосфорилирования. Эти процессы и являются механизмами активации и деактивации белков сигнального каскада.
Началом сигнального пути является внеклеточный домен рецептора эпидермального фактора роста (EGFR, Epidermal Growth Factor Receptor), связанный с тирозинкиназой, которая при связывании лиганда с рецептором активирует фосфорилирование тирозиновых остатков внутриклеточного домена рецептора EGFR (рис. 1). Кроме EGFR, в качестве рецептора могут выступать рецептор тирозинкиназы Trk A/B, рецептор фактора роста фибробластов FGFR (Fibroblast Growth Factor Receptor) и рецептор тромбоцитарного фактора роста PDGFR (Platelet-18 Derived Growth Factor receptors). С фосфорилированными тирозиновыми остатками рецептора взаимодействует белок GRB2 (Growth factor receptor-bound protein 2) посредством домена SH2 (Src Homology 2) [11].
Далее GRB2 взаимодействует посредством домена SH3 с белком SOS (Son of Sevenless) из семейства GEF (Guanine nucleotide Exchange Factor). Комплекс GRB2 и SOS приводят к активации последнего [12]. Активированный SOS способствует диссоциации ГДФ от белков семейства Ras, в том числе KRAS. К семейству RAS относятся четыре белка, кодируемые тремя генами: NRAS, HRAS, KRAS (4A и 4B). Далее RAS, являясь ГТФазой, связывает ГТФ и принимает активную форму. Активированный RAS затем активирует белок серин-треониновую киназу RAF (Rapidly Accelerated Fibrosarcoma), в том числе BRAF [13]. RAF киназа фосфорилирует и активирует киназу MEK (Mitogen-activated protein kinase kinase), которая в свою очередь активирует MAPK. MAPK может фосфорилировать и активировать многих последующих посредников передачи сигнала. Например,
MAPK активирует RSK (40S ribosomal protein S6 kinase), регулируя трансляцию мРНК, или активирует факторы транскрипции, такие как c-Myc, CREB (cAMP response element-binding protein). Все эти процессы в итоге запускают клеточную пролиферацию, ангиогенез и выживание клеток.
RAS – малая ГТФаза, которая присутствует во всех клетках организма. Состоит из шести β -цепей и пяти α -спиралей, имеет G-домен, состоящий из 166 аминокислот и принимающий непосредственное участие в связывании ГТФ и ГДФ; и С-домен, имеющий сродство к мембране. Этот компонент RAS-RAF-MEK-ERK сигнального пути выполняет функцию «переключателя» сигнального каскада, переходя из неактивной формы в активную и обратно. Этот переход сопровождается гидролизом ГТФ (активная форма) – ГДФ (не активная форма). Однако сам по себе RAS имеет очень низкую ГТФазную активность и нуждается в катализаторе, которым является белок GAP, значительно увеличивающий гидролиз ГТФ. Этот белок имеет петлю, в составе которой находится аминокислота Arg789, которая и взаимодействует с β -фосфатом ГТФ, ускоряя гидролиз. Важное значение в процессе гидролиза имеют аминокислотные остатки Gly12, Gly13 и Gln61 белка RAS. Gly12 и Gly13 на прямую не принимают участие в гидролизе, но атом водорода R-группы этих аминокислотных остатков занимают положение около каталитической петли GAP белка. И если в результате мутации происходит замена аминокислоты, то это приводит к стерическим затруднениям, вследствие которых каталитическая петля GAP белка не может занять нужную позицию. Это приводит к снижению скорости гидролиза ГТФ, вследствие чего белок RAS дольше остается связанным с ГТФ в активированном состоянии. Аминокислота белка RAS Gln61 хотя и находится далеко от места связывания ГТФ, но исполняет функцию активации молекулы воды как первичного нуклеофила [14–17]. Таким образом, мутации аминокислотных остатков в позиции 12, 13 и 61 приводят к снижению ГТФазной активности RAS [18].
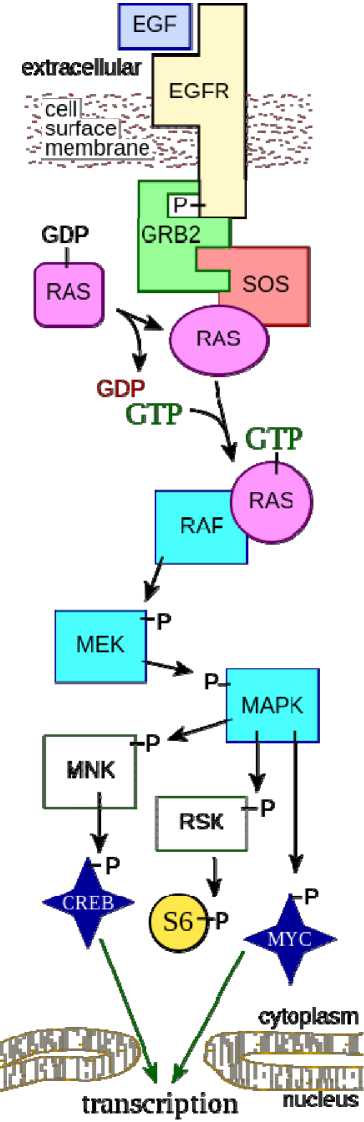
Рис. 1. Схема RAS-RAF-MEK-ERK сигнального пути. Р обозначает фосфорилирование,
Fig. 1. Scheme of the RAS-RAF-MEK-ERK signaling pathway. P stands for phosphorylation,
Семейство RAF белков представляет собой три серин-треониновые протеин-киназы: A-RAF, B-RAF, RAF-1. Киназа BRAF состоит из 766 аминокислот, образующих три консервативных домена: CR1, CR2, CR3 (Concerved Region). Домен CR3 являет- ся каталитическим центром, домен CR1 является аутоингибитором CR3 [19], а домен CR2 связывает их между собой. CR3 состоит из аминокислотных остатков 457-717, из которых формируются 2 участка: N-участок, связывающий АТФ, и С-участок, связывающий субстратные белки и состоящий из аминокислотных остатков 535-717 [20]. N-участок в своем составе имеет Р-петлю, которая стабилизирует фосфатную группу АТФ при его связывании. С-участок содержит активационную петлю, состоящую из аминокислотных остатков 596-600, функция которой заключается в блокировании киназы в неактивном состоянии, пока Р-петля вновь не свяжет молекулу АТФ [21]. Большинство из обнаруженных на сегодняшний день более 30 мутаций RAF, в том числе мутации 600 кодона гена BRAF, сосредоточены в области Р-петли, N-участка и фланкирующих районах [21]. Эти мутации нарушают взаимодействие Р-петли и активационной петли С-участка и, как следствие, приводят к отсутствию «блокировки» RAF киназы в неактивном состоянии и поэтому происходит постоянная активация этого белка. В целом EGFR представляет собой трансмембранный гликопротеин с молекулярной массой 170 кДа, состоящий из внеклеточного домена с двумя богатыми цистеином областями, трансмембранного домена и внутриклеточного домена, обладающего тирозинкиназной активностью [22].
В роли лигандов EGFR наиболее часто выступают ростовые факторы EGF и фактор некроза опухоли-a (TGF-a), а также ам-фирегулин, эпирегулин, HB-EGF и b-целлюлин. Они взаимодействуют с рецептором, вызывая его гомодимеризацию (связывание лигандом двух идентичных рецепторов) или гетеродимеризацию (связывание мономера EGFR с другим представителем семейства, например, HER2 или HER3) [23–25]. В результате происходит активация тирозинкиназы во внутриклеточном домене с последующим аутофосфорилированием рецептора и инициацией каскадов сигнальной трансдукции RAS-RAF-MEK-ERK сигнального пути, участвующего в процессе пролиферации и опухолевой прогрессии (активация инвазии, метастазирования, включение антиапоптозных механизмов) [10].
Таким образом, каскад (RAS-RAF-MEK-ERK путь) является путем, который регулирует клеточную пролиферацию, клеточный цикл и миграцию клетки. При развитии рака у человека мутации семейства RAS/RAF наиболее часто являются причиной нарушения регуляции трансдукции сигнала через этот путь. Согласно современным данным около трети всех злокачественных новообразований ассоциированы с мутациями в генах семейства RAS. Однако частоты мутаций этих генов значительно варьируют в зависимости от определенного типа рака: активирующие мутации KRAS часто обнаруживаются при немелкоклеточном раке легкого (15–20 %), раке толстой кишки (40 %) и аденокарциноме поджелудочной железы (95 %); NRAS-мутации выявляются с высокой частотой в гематологических злокачественных новообразованиях (20–30 %) [26].
Гены семейства RAS (HRAS, KRAS и NRAS) являются клеточными протоонкогенами, кодирующими белки с молекулярной массой 21kDa [27], которые принадлежат к суперсемейству малых гуанозинтрифосфатаз (ГТФаз). На уровне аминокислотной последовательности белки семейства RAS имеют высокую степень гомологии (приблизительно 80 %) и обладают способностью связывать и осуществлять гидролиз гуаниловых нуклеотидов [28]. Эти белки принимают сигналы с поверхности клеток и передают их внутрь клеток, регулируя клеточный ответ на внешние стимулы посредством активации сигнальных путей, включая RAF-MAPК, PI3К и Ral-GEF (Ral-GDS) [29].
Сигнальные пути, активируемые белками RAS, участвуют в регуляции таких клеточных процессов, как пролиферация, рост, дифференцировка, миграция, апоптоз и выживание [30].
Установлено, что соматические точко-вые мутации генов RAS, приводящие к замене аминокислот в 12-й, 13-й или 61-й позициях, присутствуют в 20–30 % злокачественных опухолей человека [31]. Такие молекулярные изменения обусловливают нарушение ГТФазной активности белка
RAS, что проявляется в его конститутивном активном состоянии. Последнее в свою очередь ведет к активации нисходящих сигнальных каскадов даже в отсутствие внеклеточных стимулов и, как следствие, к злокачественной трансформации клеток.
При анализе генома пациентов с КРР очень часто выявляются мутации (частота до 40 %) [32]. KRAS является геном, кодирующим один из белков, играющих важную роль в сигнальной системе рецептора эпидермального фактора роста (EGFR), а также регулирует белки, находящиеся далее в сигнальной системе EGFR, которые связаны с выживаемостью опухоли, ангиогенезом, пролиферацией и метастазированием [33].
При метастатическом колоректальном раке почти 60 % пациентов имеют нормальную сигнальную систему EGFR и дикий тип гена KRAS; остальные 40 % имеют мутантный тип гена [32].
NRAS также относится к семейству онкогенов RAS, ген которого расположен на первой хромосоме [29]. NRAS отличается от KRAS концевым участком белковой молекулы, что определяет его отличие в транспортировке, во внутриклеточном расположении и функции [34]. Мутация гена NRAS встречается в 3–5 % случаях КРР, чаще в кодоне 61. Мутации KRAS и NRAS взаимоисключающие [34, 35]. Наличие мутации в гене NRAS определяет наличие резистентности к анти-EGFR-терапии КРР [35–37].
Мутации в гене KRAS в опухолях толстой кишки встречаются в 30–60 % случаев. Наиболее часто мутации KRAS определяются в экзоне 2, кодонах 12 и 13. Однако описаны мутации в экзоне 3, кодоне 61 и в экзоне 4, кодонах 117 и 146. Мутации в гене NRAS (в идентичных экзонах и кодонах) при КРР составляют до 5 % [38].
Хирургическое лечение КРР играет главную роль в излечении больных, однако пациенты с III стадией рака нуждаются в адъювантной химиотерапии (АХТ), что было про-демонcтрировано во множестве рандомизированных исследований. Таргетная терапия, по сравнению с конвенциональной химиотерапией, имеет ряд преимуществ: индивидуализация назначения, более низкая токсичность, таблетированные формы большинства препаратов исключают необходимость госпитализации и позволяют больным радикально не менять образ жизни.
В качестве основных мишеней целенаправленной терапии могут выступать многочисленные элементы сигнальных путей, связанные с регуляцией клеточного цикла и апоптоза, нарушение которых ассоциировано со злокачественным ростом. Рецептор эпидермального фактора роста (EGFR), или HER1, – трансмембранный гликопротеин с молекулярной массой 170 kD, обладающий тирозинкиназной активностью, является наиболее хорошо изученной мишенью.
Основные механизмы активации EGFR-зависимых сигнальных путей в опухолевых клетках обеспечиваются: мутацией тирозинкиназного домена гена EGFR и, как следствие этого, его аутоактивацией при отсутствии факторов роста, приводящей к неконтролируемой пролиферации; гиперэкспрессией EGFR; избыточной продукцией факторов роста – лигандов EGFR (TGF-a, EGF) [38].
Существует несколько вариантов блокирования онкогенного эффекта, реализуемого через активированный EGFR: 1) использование низкомолекулярных ингибиторов, способных воздействовать на внутриклеточный, несущий мутацию, домен EGFR, и прерывать процесс тирозинкиназного фосфорилирования; 2) применение рекомбинантных пептидных лигандов EGF и/или TGF-a, конъюгированных с проникающими внутрь клетки цитотоксинами; 3) использование моноклональных антител, связывающих экстрацеллюлярный участок рецептора или образующих неактивный комплекс с его лигандами EGF и TGF-a. В настоящее время к клиническому применению разрешены девять ингибиторов передачи сигнала в клетки (иматиниб, сунитиниб, сорафениб, лапати-ниб, гефитиниб, эрлотиниб, дазатиниб, ни-лотиниб, пазопаниб) и пять моноклональных антител (трастузумаб, ритуксимаб, бева-цизумаб, цетуксимаб, панитумумаб) [39].
Таким образом, для лечения больных КРР успешно применяются препараты на основе моноклональных антител к EGFR, которые, связываясь с ним на поверхности клетки, блокируют запуск сигнального каскада и таким образом препятствуют росту и развитию опухоли [40]. Эффективность лечения этими препаратами зависит от молекулярногенетических изменений в опухоли: статуса EGFR, наличия мутаций в других участниках сигнального каскада – онкогенах KRAS и BRAF – и некоторых других факторов. При отсутствии мутаций в гене KRAS эффективность лечения РТПК очень высока, увеличивается средняя продолжительность жизни, уменьшается количество рецидивов. В то же время при наличии в клетках опухоли активирующих мутаций в гене KRAS использование анти-EGFR антител не приводит к положительным результатам [41]. В 30–40 % случаев РТПК выявляются следующие мутации в 12 и 13 кодонах KRAS (85–90 %), которые коррелируют с резистентностью опухолей к анти-EGFR терапии: G12C, G12S, G12R, G12A, G12V, G12D, G13D. Значительно реже выявляются мутации в кодонах 61 (5 %) и 146 (5 %) [41, 42]. Таким образом, тест на наличие мутации гена KRAS необходим пациентам с РТПК для оценки возможности применения таргетной терапии анти-EGFR антителами [41, 42].
Активирующие мутации BRAF встречаются, по разным данным, в 5–15 % случаев РТПК, в 95 % случаев это мутация V600E
[43–46]. Существуют противоречивые данные о предсказательной роли мутации BRAF V600E в отношении ответа опухоли на анти-EGFR терапию и прогностической значимости в отношении прогрессирования заболевания [47, 48]. В настоящее время имеются немногочисленные исследования частоты мутаций в гене KRAS и одно исследование мутаций в гене BRAF при РТПК у российских пациентов [43–46].
Доказано, что активация генов семейства RAS, за счет мутаций сводит на нет эффект ингибирования EGFR моноклональными антителами при терапии мета-стазированного колоректального рака (мКРР) [49–52].
Таким образом, эффективность тар-гетной терапии при КРР зависит от статуса EGFR, наличия мутаций в онкогене KRAS и некоторых других факторов. При отсутствии мутаций в гене KRAS замедляется прогрессия заболевания (9,6 против 8,0 мес.) и увеличивается общая продолжительность жизни больного (23,9 против 19,7 мес.) [53]. В тоже время, в случае наличия активирующих мутаций в гене KRAS в клетках опухоли больного, использование анти-EGFR антител не приводит к положительным результатам [41, 42].
Таким образом, современные возможности ранней диагностики РТК, прогнозирования течения заболевания, определения чувствительности/резистентности к лекарственным препаратам основаны на индивидуальных наследственных и соматических молекулярно-генетических характеристиках.
Список литературы Молекулярно-генетические механизмы сигнального каскада RAS-RAF-MEK-ERK, связанные с развитием опухолевого процесса и назначением таргетных препаратов при колоректальном раке
- Zemlyanoy VP, Trofimova TN, Nepomniachtchi SL, Dementieva TV. Practical Oncology. 2005;6(2):71-81.
- Gorbunova V.A. Research data for CRAS- and RAS-unmutated (wild) type of colorectal cancer. Oncological coloproctology. 2015;1:26-35.
- Davydov M.I., Aksel E.M. Statistics of malignant neoplasms in Russia and the CIS countries in 2012. Vestnik RONC im. N.N. Blokhina = Bulletin of N.N. Blokhin Russian Cancer Research Center 2014. (In Russ).
- Pospekhova N.I., Shubin V.P., Tsukanov A.S., Frolov S.A., Shelygin Yu.A. Molecular genetic markers in on-cocoloproctology: assistance in diagnosis, prognosis, treatment. Clinical laboratory diagnostics. 2014;9:46-47. (In Russ).
- Starinsky V.V., Petrova G.V., Chissov V.I. The incidence of malignant neoplasms in the population of Russia in 2000. Ros OnkolZhurn. 2002;3:39-44. (In Russ).
- Pasevich D.M., Sushkov S.A., Semenov V.M. Molecular genetic aspects of malignant neoplasms of the colon. News of surgery. 2016;24(2):184-192.
- Brovkina O.I., Gordiev M.G., Khodyrev D.S., Nikitin A.G., Averyanov A.V. Use of aberrantly methylated SEPT9 and VIM genes for clinical diagnosis of colorectal cancer Clinical practice No. 4, 2016.
- Schneider K.U., Dietrich D., Fleischhacker M. et al. Correlation of SHOX2 Gene Amplification and DNA Meth-ylation in Lung Cancer Tumors. BMC Cancer. 2011 Mar 22;11:102. https://doi.org/10.1186/1471-2407-11-102.
- Mikeska T., Craig J.M. DNA Methylation Biomarkers: Cancer and Beyond. Genes. 2014;5:821 -864.
- Porebska I., Harlozinska A., Bojarowski T. Expression of the tyrosine kinase activity growth factor receptors (EGFR., ERB B2., ERB B3) in colorectal adenocarcinomas and adenomas. Tumour Biol. 2000;21(2):105-115.
- Chulze, W.X., Deng, L., Mann, M. Phosphotyrosine interactome of the ErbB-receptor kinase family. Molecular systems biology. 2005;1 (1):2005-2008.
- Zarich, N., Oliva, J.L., Martínez, N., Jorge, R., Ballester, A., Gutiérrez-Eisman, S., García-Vargas, S., Rojas, J.M. Grb2 is a negative modulator of the intrinsic Ras-GEF activity of hSos1. Molecular Biology of the Cell. 2006;17(8):3591-3597.
- Avruch, J., Khokhlatchev, A., Kyriakis, J.M., Luo, Z., Tzivion, G., Vavvas, D., Zhang, X.F. Ras activation of the Raf kinase: tyrosine kinase recruitment of the MAP kinase cascade. Recent Progress in Hormone Research. 2001;56(1):127-155.
- Fraser, J.S. van den Bedem, H., Samelson, AJ, Lang, PT, Holton, JM, Echols, N., Alber, T. Accessing protein conformational ensembles using room-temperature X-ray crystallography. Proc Natl Acad Sci US A. 2011;108(39):16247-16252.
- Frech, M., Darden, TA, Pedersen, LG, Foley, CK, Charifson, PS, An-derson, MW, Wittinghofer, A. Role of glutamine-61 in the hydrolysis of GTP by p21H-ras: an experimental and theoretical study. Biochemistry. 1994;33(11):3237-3244.
- Ghosh, A., Praefcke, G.J., Renault, L., Wittinghofer, A., Herrmann, C. How guanylate-binding proteins achieve assembly-stimulated processive cleavage of GTP to GMP. Nat Cell Biol. 2006;440(7080):101-104.
- Goitre, L., Trapani, E., Trabalzini, L., Retta, S.F. The Ras superfamily of small GTPases: the unlocked secrets. Methods Mol Biol. 2014;1120:1-18.
- Stolze, B., Reinhart, S., Bulllinger, L., Frohling, S., Scholl, C. Comparative analysis of KRAS codon 12, 13, 18, 61, and 117 mutations using human MCF10A iso-genic cell lines. Sci Rep. 2015;5:8535.
- Cutler, R.E. Jr, Stephens, R.M., Saracino, M.R., Morrison, D.K. Autoregu-lation of the Raf-1 serine / threonine kinase. PNAS. 1998;95(16):9214-9219.
- Hanks, S.K., Hunter, T. Protein kinases 6. The eukaryotic protein kinase superfamily: kinase (catalytic) domain structure and classification. FASEB J. 1995;9(8):576-596.
- Wan, PT, Garnett, MJ, Roe, SM, Lee, S., Niculescu-Duvaz, D., Good, VM, Jones, CM, Marshall, CJ, Springer, CJ, Barford, D., Marais, R Cancer Ge-nome Project. Mechanism of activation of the RAF-ERK signaling pathway by onco-genic mutations of B-RAF. Cell. 2004;116(6):855-867.
- Downward, J., Parker, P., Waterfield, M.D. Autophosphorylation sites on the epidermal growth factor receptor. Nature. 1984;311:483-485.
- Coffey, R.J. Jr, Goustin, A.S., Soderquist, A.M., Shipley, G.D., Wolfshohl, J., Carpenter, G., Moses, H.L. Transforming growth factor alpha and beta expression in human colon cancer lines: implications for an auto-crine model. Cancer. 1987;47:4590-4594.
- Carpenter, G., Cohen, S. Epidermal growth factor. J Biol Chem. 1990;265:7709-7712.
- Messa C., Russo F., Caruso M.G., Di Leo A. EGF, TGF-a, and EGFR in human colorectal adenocarcinoma. Acta Oncol. 1998;37:285-289.
- Grant, S., Qiao, L., Dent, P. Roles of erbB family receptor tyrosine kinases, and downstream signaling pathways, in the control of cell growth and survival. Front Biosci. 2002;7:376-389.
- Smal M.P., Rolevich A.I., Nabebina T.I., Krasny S.A., Goncharova R.I. Opposite associations of HRAS and KRAS gene mutations with clinical parameters of bladder cancer. Vavilov Journal of Genetics and Breeding. 2015;19(5):638-646.
- Yan Z., Chen M., Perucho M., Friedman E. Oncogenic Ki-ras but not oncogenic Ha-ras blocks integrin be-ta1-chain maturation in colon epithelial cells. Biol. Chem. 1997;272(49):30928-30936. https://doi.org/10.1074/jbc.272.49.30928
- Castellano E, Santos E. Functional specificity of Ras isoforms: sosimilar but so different. Genes Cancer. 2011;2(3):216-231. https://doi.org/10.1177/194760191140808
- Malumbres M., Barbacid M. RAS oncogenes: the first 30 years. Nat. Rev. Cancer. 2003;3(6):459-465. https://doi.org/10.1038 / nrc1097
- Pollard C., Smith S.C., Theodorescu D. Molecular genesis of non-mus cle-invasive urothelial carcinoma (NMIUC). Expert. Rev. Mol. Med. 2010;12:e10. https://doi.org/10/1017/S1462399410001407
- Forbes SA, Beare D., Gunasekaran P., Leung K., Bindal N., Boutse lakis H., Ding M., Bamford S., Cole C., Ward S., Kok CY, Jia M., De T ., Teague JW, Stratton MR, McDermott U., Campbell PJ COSMIC: exploring the world's knowledge of somatic mutations in human cancer Nuc. Acids Res. 2015;43:D805-811. https://doi.org/10.1093/nar/gku1075
- Linardou H., Briasoulis E., Dahabreh I. J., et al. All about KRAS for clinical oncology practice: gene profile, clinical implications and laboratory recommendations for somatic mutational testing in colorectal cancer. Cancer Treat. Rev. 2011;37:221-233.
- Ying H.-Q., Wang F., He B.-S., et al. The involvement of Kras gene 3'-UTR polymorphisms in risk of cancer and influence on patient response to anti-EGFR therapy in metastatic colorectal cancer: a metaanalysis. On-coTargets Ther. 2014;7:1487-1496.
- Haigis K. M., Kendall K. R., Wang Y. et al. Differential effects of oncogenic K-Ras and N-Ras on proliferation, differentiation and tumor progression in the colon. Nat Genet. 2008;40:600-8.
- De Roock W., Claes B., Bernasconi D. et al. Effects of KRAS, BRAF, NRAS and PIK3CA mutations on the efficacy of cetuximab plus chemotherapy in chemotherapy-refractory metastatic colorectal cancer: a retrospective consortium analysis. Lancet Oncol. 2010;11:753-62.
- Seymour M.T., Brown S.R., Richman S. et al. Addition of panitumumab to irinotecan: results of PICCOLO, a randomized controlled trial in advanced colorectal cancer (aCRC). J Clin Oncol. 2011 ;29 (Suppl.) [Abstract 3523].
- Oliner K., Peeters M., Siena S. et al. Evaluation of the gene mutations beyond KRAS as predictive bi-omarkers or response to panitumumab in a randomized, phase III monotherapy study of metastatic colorectal cancer (mCRC). J Clin Oncol. 2011;29 (Suppl.) [Abstract 3530].
- Gervas P.A., Litvyakov N.V., Popova N.O. et al. Cherdyntseva Problems and prospects of improving molecular genetic diagnostics for the appointment of targeted drugs in oncology. Siberian Journal of Oncology. 2014;2(62):46-55.
- Protsenko S.A. Targeted therapy for melanoma, gastrointestinal stromal tumors, protuberance dermatofibro-sarcoma. Practical Oncology. 2010;11(3):162-170.
- Jean G.W., Shah S.R. Epidermal growth factor receptor monoclonal antibodies for the treatment of metastatic colorectal cancer. Pharmacotherapy. 2008;28(6):742-754. https://doi.org/10.1592/ phco.28.6.742
- Lièvre A., Bachet JB, Le Corre D. et al. KRAS mutation status is predictive of response to cetuximab therapy in colorectal cancer. Cancer Res. 2006;66(8):3992-3995.
- Van Krieken JH, Jung A., Kirchner T. et al. KRAS mutation testing for predicting response to anti-EGFR therapy for colorectal carcinoma: proposal for an European quality assurance program. Virchows Arch. 2008;453(5):417-431. https://doi.org/10.1007/s00428-008-0665-y
- Kit O.I., Vodolazhsky D.I., Dvadnenko K.V. et al. Frequency of mutations in the KRAS gene in various clinical groups of patients with colorectal cancer in the south of Russia. Medical genetics. 2014;12(150):35-41.
- Mazurenko N.N., Gagarin I.M., Tsyganova I.V. et al. Frequency and spectrum of KRAS mutations in metastatic colorectal cancer. Problems of Oncology. 2013;59(6):751-755.
- Shubin V.P., Pospekhova N.I., Tsukanov A.S. et al. Frequency and spectrum of mutations in the KRAS gene in colon cancer of different localization and cancer of the anal canal. Medical Genetics. 2014;5(143):31-35.
- Yanus GA, Belyaeva AV, Ivantsov AO et al. Pattern of clinically relevant mutations in consecutive series of Russian colorectal cancer patients. Med. Oncol. 2013;30(3):686. https://doi.org/10.1007/s12032-013-0686-5.
- Di Nicolantonio F., Martini M., Molinari F. et al. Wild -type BRAF is required for response to panitumumab or cetuximab in metastatic colorectal cancer. J. Clin. Oncol. 2008;26(35):5705-5712. https://doi.org/10.1200/ JCO.2008.18.0786
- Laurent-Puig P., Cayre A., Manceau G. et al. Analysis of PTEN, BRAF, and EGFR status in determining benefit from cetuximab therapy in wild-type KRAS metastatic colon cancer. J. Clin ... Oncol. 2009;27(35):5924-5930. https://doi.org/10.1200 / JCO.2008.21.6796
- Imyanitov E.N. Clinical and molecular aspects of colorectal cancer: etiopathogenesis, prevention, individualization of treatment. Practical Oncology. 2005;6(2):65-68.
- De Roock W., Jonker DJ, Di Nicolantonio F. et al. Association of KRAS p.G13D mutation with outcome in patients with chemotherapy-refractory metastatic colorectal cancer treated with cetuximab. JAMA. 2010; 304(16):1812-1820. https://doi.org/10.1001/jama.2010.1535
- Roth AD, Tejpar S., Delorenzi M. et al. Prognostic role of KRAS and BRAF in stage II and III resected colon cancer: results of the translational study on the PETACC-3, EORTC 40993, SAKK 60 -00 trial. J. Clin. Oncol. 2010;28(3):466-474. https://doi.org/ 10.1200 / JC0.2009.23.3452
- Tie J., Lipton L., Desai J. et al. KRAS mutation is associated with lung metastasis in patients with curatively resected colorectal cancer. Clin. Cancer Res. 2011; 17(5): 1122-1130. https://doi.org/10.1158/1078-0432. CCR-10-1720
- Douillard J.Y., Zemelka T., Fountzilas G. et al. F0LF0X4 with cetuximab vs. UFOX with cetuximab as first-line therapy in metastatic colorectal cancer: The randomized phase II FUTURE study. Clin Colorectal Cancer. 2014;13(1):14-26.
