Молекулярное моделирование, докинг и моделирование молекулярной динамики для изучения взаимодействия бел-ков PAD4 и EDS1 у Solanum lycopersicum L.

Автор: Tandon G., Prasad S.S., Singh A., Chester J.R.E, Singh P.K., Kaur S., Jaiswal S., Iquebal M.A., Rai A., Kumar D., Singh S.
Журнал: Сельскохозяйственная биология @agrobiology
Рубрика: Молекулярное моделирование
Статья в выпуске: 1 т.61, 2026 года.
Бесплатный доступ
У растений биотический стресс становится одним из основных препятствий для производства продуктов питания, поэтому важно понимать, какие метаболические пути и гены связаны с системным иммунитетом. Enhanced Disease Susceptibility 1 (EDS1) и Phytoalexin Lacking 4 (PAD4) — два важных белка, которые необходимы для накопления салициловой кислоты. Это, в свою очередь, контролирует экспрессию белков, связанных с патогенезом (PR-белков), которые активируют защитные механизмы против атак патогенов. Вычислительные методы позволяют прогнозировать основные белок-белковые взаимодействия и ключевые гены, вовлеченные в сигнальные пути. В настоящем исследовании мы провели 3D-моделирование и изучили взаимодействие EDS1 и PAD4 у томата (Solanum lycopersicum), используя метод гомологического моделирования. Предсказанные модели были дополнительно валидированы, после чего для них провели докинг белок-белковых взаимодействий с использованием HADDOCK. Сравнительный анализ пространственных структур белков и потенциальных взаимодействий выявил 13 водородных связей и 5 гидрофобных взаимодействий между белками. Для наблюдения за динамическим поведением белков было выполнено молекулярно-динамическое моделирование (МДМ) в течение 50 нс с использованием GROMACS. При анализе результатов МДМ учитывали различные параметры, включая среднеквадратичное отклонение (RMSD), потенциальную энергию, а также использовали метод главных компонент (PCA). Докинг-анализ выявил наличие комплексов EDS1 и PAD4 у томатов, тем самым подтвердив, что салициловая кислота может быть вовлечена в систему защиты растения. Гены салицилатного сигнального пути могут быть непосредственно использованы в дальнейших исследованиях, необходимых для разработки новых сортов.
Solanum lycopersicum, биосинтез салициловой кислоты, EDS1, PAD4, системная резистентность, R-ген, молекулярно-динамическое моделирование, гомологическое моделирование, докинг белков
Короткий адрес: https://sciup.org/142247331
IDR: 142247331 | УДК: 635.64:58.071:577.29:51-76 | DOI: 10.15389/agrobiology.2026.1.72rus
Molecular modelling, docking and dynamics simulations of Solanum lycopersicum PAD4 using EDS1 proteins
As biotic stress is one of the major impediments to food production, it is important to decipher systemic immunity-related pathways and genes. Enhanced Disease Susceptibility 1 (EDS1) and Phytoalexin Lacking 4 (PAD4) are two important proteins that together are essential for the accumulation of salicylic acid (SA). This accumulation, in turn, controls the expression of pathogenesis-related (PR) proteins, which in turn activates defense pathways against pathogen attacks. A computational approach can predict major protein-protein interactions and major signaling pathways possessing key genes. In this study, we conducted 3D modeling and examined the interaction of EDS1 and PAD4 in tomatoes (Solanum lycopersicum), using a homology modeling approach. The predicted models were further validated and then subjected to protein-protein docking using HADDOCK. Based on a comparative analysis of the spatial structures of proteins and potential interactions were revealing the presence of 13 hydrogen bonds and 5 hydrophobic interactions between both the proteins. Molecular dynamics simulations (MDSs) for 50ns were performed using GROMACS to observe the dynamic behavior of the proteins. Various parameters, including RMSD, potential energy, and PCA, were considered during MDS analysis. Docking analysis revealed the presence of EDS1 and PAD4 complexes in tomatoes, thus supporting the possibility that SA might operate as a defense pathway. Such pathway genes can be directly targeted for further studies required for new variety development.
Текст научной статьи Молекулярное моделирование, докинг и моделирование молекулярной динамики для изучения взаимодействия бел-ков PAD4 и EDS1 у Solanum lycopersicum L.
Растения в силу неподвижного образа жизни подвержены различным видам экологического стресса, что существенно влияет на рост и урожайность сельскохозяйственных культур. Стрессы можно разделить на биотические и абиотические. Абиотический стресс вызывают засуха, засоление, низкие и высокие температуры. Биотический стресс провоцируют различные патогены — грибы, бактерии, вирусы или нематоды. Стрессоры могут воздействовать на растения по отдельности или в сочетании друг с другом. Несмотря на использование улучшенных сортов и применение пестицидов для противодействия биотическим стрессам, вредители и болезни все еще вызывают потери урожая в размере 20-30 % (1).
Для преодоления стрессов растения выработали сложные механизмы, с помощью которых реагируют на атаки патогенов. Эта устойчивость в основном контролируется взаимодействием по принципу «ген-на-ген» (gene-for-gene), происходящим между генами устойчивости растения (resistance, R ) и генами авирулентности (avirulence, Avr ) патогена (2). Распознавание Avr активирует реакцию гиперчувствительности (hypersensitive response, HR), и эта HR, в свою очередь, приводит к гибели клеток в месте проникновения патогена или вблизи него (3). Считается, что HR ограничивает распространение патогенов от места заражения (4). При участии R -гена активируется сигнальный путь, зависящий от салициловой кислоты (salicylic acid, SA), что вызывает экспрессию PR (pathogenesis-related) белков. Сообщалось о других (помимо SA-зависимого пути) защитных реакциях растений, включая пути,
∗ При финансировании от Индийского совета сельскохозяйственных исследований (ICAR) при правительстве Индии в рамках создания Центра передовых суперкомпьютерных вычислений ASHOKA в ICAR-IASRI, Нью-Дели. Частично работа поддержана программой CABin Scheme (код проекта: 1004936) ICAR-IASRI, Нью-Дели.
зависимые от этилена (ethylene, ET) или жасмоновой кислоты (jasmonic acid, JA). Также высказано предположение, что SA-зависимый путь активируется в ответ на атаку биотрофов, тогда как пути ET и JA — некротрофов (5).
Для правильного накопления SA необходимы два белка — Enhanced Disease Susceptibility 1 (EDS1) и Phytoalexin Lacking 4 (PAD4) (6). EDS1 взаимодействует с PAD4 и Senescence Associated Gene 101 (SAG101). Белки EDS1 и PAD4 необходимы для инициации HR. EDS1 и PAD4 присутствуют как в ядре, так и в цитоплазме, тогда как SAG101 локализован исключительно в ядре (7). EDS1, PAD4 и SAG101 структурно принадлежат к семейству липазо-/эстеразоподобных белков (8). EDS1 играет ключевую роль в обеспечении резистентности, которая опосредована большим подклассом внутриклеточных нуклеотид-связывающих рецепторов с обогащенными лейцином повторами (nucleotide-binding leucine-rich repeat, NLR), обозначаемых TNL и содержащих N-концевой домен Toll/интерлейкин-1 рецептора (Toll/interleukin 1 receptor, TIR) ( 9). Эти TNLs распознают эффекторы патогенов и индуцируют эффектор-активируемый иммунитет (effector-triggered immunity, ETI), который усиливает транскрипционные программы базовой иммунной системы, а также связан с локальной запрограммированной гибелью клеток хозяина. Таким образом, EDS1 участвует в усилении гиперчувствительных реакций растений (10).
Функциональные ортологи EDS1 и PAD4 были идентифицированы у многих покрытосеменных растений, включая рис и виноград, то есть комплекс EDS1 представляет собой древнюю систему защиты растений, существовавшую до появления ETI (11, 12). Взаимодействие EDS1-PAD4 для обеспечения устойчивости и их роль в SA-зависимом защитном пути также описаны у Arabidopsis thaliana (6). Взаимосвязь EDS1-PAD4 не только способствует биосинтезу SA, но и играет важную роль во врожденном иммунитете, усиливая SA-зависимую резистентность у Arabidopsis thaliana (13).
В настоящее время последовательности обоих белков (EDS1 и PAD4) томатов доступны в NCBI, но информация об их структуре отсутствует. Более того, до сих пор не изучено их взаимодействие, служащее частью SA-зависимого пути в зашитой реакции растений
Томат ( Solanum lycopersicum ) служит модельной системой для изучения развития плодов. Также это богатый источник множества питательных веществ, таких как β -каротин, ликопин, флавоноиды, витамины A, B и C, производные гидроксикоричной кислоты и минералы (фосфор, калий, магний и др.), он считается важной овощной культурой (14, 15). Мировое производство и потребление этой пасленовой культуры постоянно растет (16). Однако до 70 % урожая томата теряется из-за различных биотических и абиотических стрессов. Биотические агенты, такие как бактерии, вирусы, грибы или нематоды, становятся причиной примерно 200 заболеваний и вызывают потерю урожая томатов на 90 % (17). Таким образом, изучение комплекса EDS1-PAD4 может быть полезно для раскрытия SA-зависимых механизмов ответа на биотический стресс у томата.
В настоящей работе предпринята попытка изучить взаимодействие EDS1 и PAD4 посредством построения их трехмерных моделей, использования гомологического моделирования для понимания молекулярных механизмов на атомном уровне, а также анализа взаимодействия белков методом докинга. После этого были изучены паттерны стабильности с помощью моделирования молекулярной динамики (МД) в течение 50 нс.
Главная цель исследования заключалась в выявлении изменений, происходящих в EDS1 и PAD4 при их взаимодействии.
Ìåòîäèêà. Получение последовательностей и анализ кон- сервативных доменов. Аминокислотные последовательности EDS1 и PAD4 для Solanum lycopersicum и Arabidopsis thaliana были получены из базы данных белков NCBI. Домены обоих белков у S. lycopersicum были идентифицированы с помощью инструментов предсказания доменов, включая CD-Search, SMART (18) и Interproscan (12). Впоследствии было выполнено попарное выравнивание последовательностей EDS1 и PAD4 с использованием Blastp (19), доступного в NCBI (20), для определения консервативных регионов у S. lycopersicum и A. thaliana.
Молекулярное моделирование. Трехмерные структуры были построены посредством гомологического моделирования в программе Modeller 9.14 (21). Modeller использует функции плотности вероятности (probability density functions, PDFs) в качестве пространственных ограничений вместо энергии. Modeller предсказал все 10 моделей; одна модель была проверена на стереохимическую совместимость, чтобы отметить все отклонения диэдральных (двугранных) углов, нормальную связь и длину нековалентной связи между атомами. Шаблоны для EDS1 и PAD4 были найдены с помощью Blastp для сравнения с базой данных белков с целью построения моделей (22). Файлы выравнивания целевых и шаблонных белков были получены с помощью программы Clustalw . Этот файл выравнивания мы использовали в качестве входных данных в Modeller для построения трехмерных моделей. Полученные 3D-модели оценивали на основе показателя DOPE (Discrete Optimized Protein Energy) (23) и оптимизировали для структурной доработки в программе GalaxyRefine (24).
Проверка качества структуры. Уточненные структуры были проверены на стереохимическое качество с помощью сервера проверки и анализа структуры (Structure Validation and Analysis Server, SAVES), предоставляющего несколько соответствующих программ анализа. ERRAT проверяет статистику нековалентных связей между различными типами атомов. PROCHECK помогает анализировать стереохимическое качество структуры белка. VERIFY-3D оценивает совместимость атомной модели с ее собственной аминокислотной последовательностью. WHAT_IF участвует в интенсивной проверке стереохимических параметров остатка модели. Все эти программы в составе SAVES помогают в полной и надлежащей проверке стереохимии построенных моделей белков (25).
Кроме того, для оценки качества использовали сервер ProSA (26). С помощью MolProbity были рассчитаны длины связей и углы связей (19). Для статистической проверки также использовали программы VADAR, PROCESS и GeNMR (27). Общее стереохимическое качество всех моделей EDS1 и PAD4 оценивали с помощью ProQ (28) и ModFOLD V4.0 (29). После надлежащей проверки были выбраны окончательные модели, а затем проведена минимизация их энергии с помощью Swiss PDB Viewer (30) — автономной программы, используемой для анализа макромолекул.
Молекулярный докинг и анализ белок-белковых взаимодействий. С помощью сервиса LIGSITE были определены активные аминокислотные остатки обоих белков (PAD4 и EDS1), участвующие во взаимодействии между ними . LIGSITE — это эффективный по времени и автоматизированный алгоритм, способный обнаруживать карманы на поверхности белка, которые могут быть сайтами связывания. Карманы идентифицируются с помощью ряда простых операций на кубической сетке. Информацию о предсказанных активных остатках использовали для докинга с помощью HADDOCK (High Ambiguity Driven Biomolecular DOCKing)
Молекулярно-динамическое моделирование. Молекулярно-динамическое моделирование (МДМ, molecular dynamics simulations, MDSs) проводили для EDS1, PAD4 и комплекса EDS1-PAD4 с использованием пакета GROningen MAchine for Chemical Simulations (GROMACS). В GROMACS (31, 32) для построения топологий используются силовые поля CHARMm (33). Кубическая ячейка была заполнена молекулами воды TIP3P (водное окружение) с сохранением расстояния до белка 1 нм, что создавало периодические граничные условия (Periodic Boundary Conditions, PBC). При pH 7,0 были различимы разные стадии процесса протонирования для каждой ионизируемой аминокислоты. Для нейтрализации общего заряда системы при необходимости добавлялись противоионы натрия.
Энергию сольватированных структур минимизировали с использованием алгоритмов наискорейшего снижения и сопряженных градиентов, пока максимальная сила не становилась меньше 100 кДж моль - 1 нм - 1. Минимизированная структура подвергалась анализу динамики с ограничением положения течение 25 пс, при котором молекулы белка были зафиксированы и допускалось только движение молекул воды. Затем вся система (белок и растворитель) подвергалась динамическому моделированию без учета положения. Длины связей были ограничены их равновесными положениями с помощью алгоритма LINear Constraint Solver (LINCS) (34). Для поддержания температуры и давления во всех системах применяли баростат Берендсена с константами связывания соответственно 1,0 и 0,1 пс. Для расчета электростатических взаимодействий использовали метод Эвальда с сеткой зарядов с расстоянием отсечки 0,9 нм (35). Для финального анализа при длительности 50 нс при 300 К и давлении 1 атм использовали системы согласно описанию (36).
В общей сложности для каждой системы (EDS1, PAD4 и их комплекс) провели два моделирования в одинаковых условиях с помощью GROMACS одновременно с применением доступного нам суперкомпьютерного инструментария ASHOKA (Advanced Supercomputing Hub for Omics Knowledge in Agriculture; суперкомпьютерный хаб объединяет 256 вычислительных модулей, образующих кластер под управлением Linux). Все графики строили в программе Grace .
Анализ траекторий молекулярной динамики. Для анализа траектории были рассчитаны функциональные и структурные ограничения с использованием GROMACS: среднеквадратичное отклонение (root mean square deviation, RMSD), среднеквадратичная флуктуация (root mean square fluctuation, RMSF), потенциальная энергия и площадь поверхности, доступная для растворителя (solvent-accessible surface area, SASA). Площадь поверхности взаимодействия рассчитывали по формуле: Площадь поверхности взаимодействия = SASAeds1 + SASApad4 - SASAcomplex [1].
Также было рассчитано общее число остатков и атомов соответствующих остатков, взаимодействующих на границе раздела.
Анализ главных компонент. Для понимания общего характера изменения координат атомов в EDS1 и PAD4 во время МДМ был проведен анализ основных мод движения, или анализ главных компонент
(PCA). PCA уменьшает размерность сложной системы и, как следствие, характеризует как кумулятивное, так и общее движение в белковой системе. RMSF вносят наибольший вклад в PC1. Корреляция между движением частиц, прямо пропорциональная функциональной активности белка, служит причиной флуктуаций в системе. Собственные значения, которые также характеризуют общую подвижность, связанную с собственным вектором, являются суммой вариаций коллективного движения для каждого атома. Для расчетов PCA использовали встроенные функции GROMACS — g_aneig и g_covar. Мы ограничили наше исследование атомами C α , поскольку они значимо характеризуют базовые пространственные изменения и менее подвержены статистическому шуму (37).
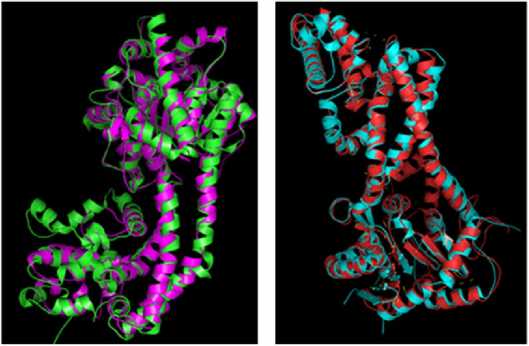
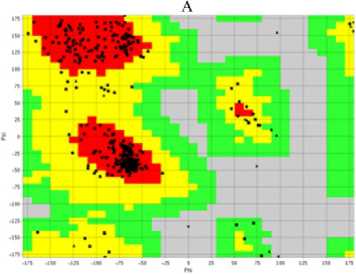
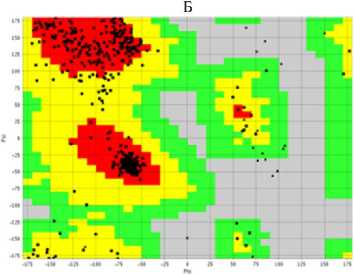
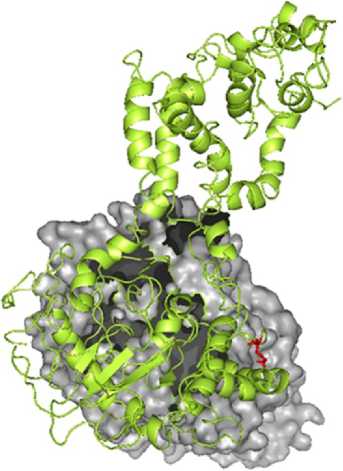
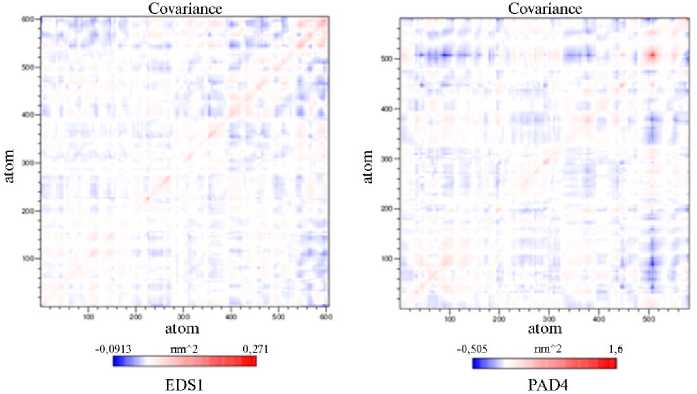
Ðåçóëüòàòû. Выявление последовательностей и анализ консервативных доменов . Структура белков EDS1 и PAD4 S. lycoper-icum доступна в NCBI (соответственно номер AAX73302.1, 602 аминокислоты, и номер XP_004231611.1, 578 аминокислот). В случае A. thaliana EDS1 (AAD20950.1) и PAD4 (OAP01954.1) содержали соответственно 623 и 541 аминокислоту. Анализ консервативных доменов, проведенный с использованием базы данных консервативных доменов, подтвердил, что EDS1 и PAD4 S. lycopersicum содержали домены семейства липаз класса 3. В EDS1 консервативный домен локализовался в положении 46VFAF…LAPL200 и имел длину 154 аминокислотных остатка; значение битового рейтинга (bit score) 76,56 и e-значение 2,30e-16; в PAD4 — в 92VHAGL…LFFA196 длиной 104 аминокислотных остатка; значение битового рейтинга (bit score) 75,41 и e-значение 4,21e-16. Выравнивание последовательностей EDS1 и PAD4 между S. lycopersicum и A. thaliana выявило разные консервативные регионы. Нуклеофильный участок GHSSGGA, образующий острый угол с основной цепью («nucleophilic elbow»), был консервативен в последовательности EDS1 у обоих видов (рис. 1). При сравнении консервативных доменов у обоих видов было обнаружено, что семейство липаз (класс 3) присутствует в обоих белках.
Query 38
Sbjct 36
Query 93
Sbjct 93
YHXEEAGAWIFAFQPSFSEKDFFDPDHKSSFGEIKIJiRVQFPC4RKIGKGDVATWiEAF 89 Y К V*FAF . 5 -- - -SFGE -W FP *R -G +VA WE F
YKKINGSTDWFAFAGTL5SDGWY---5HISFGEKEIHTTLFPSLR5VGTDEVAKVIiEVF 92
Query 13 ASVMISTPLFTDSWSSCNTAN---CN6SIKIH)IA6ITYVAIPAVSK-----IQLGNLV
A»— STPL »SW C A* CN ** * YV V • NLV
Sbjct 15 AALVASTPLLEESWKVCGVADASVGCN--FAVMRVGETAYVGFSGVKLGAGVDQSCRNLV
LKMLEAVIDPRTSFQASVE'IAVRSRKQIVF SKSSGGA A1LATWYLEKYFIR-NPIWY 148 g -hQ +«$ + VE A* *Q-VF GiSSGGA AIEA *H LE R N ♦*
ATRFEERD-KSSIKNEVEIWWIW- 6mSSG£ AILAAlHCLECCRTRPWGtn, 151
Query 64 GLPVTGOVLFPGLSSOEPLP-HVDAAILKLFLQLKIKEGLELELL—-GKKLWITGKS
LP 0 IF I (1 P P W A »L Л • I *— К wires
Sbjct 73 PIP—OELFFSLCVOGPOPAMVHAGLLHIFQSVYIMLFRDQIWEIHNTSKSIVITGHS
Query 149 LEPRCVTFGAPLVGDSIFSHALGREKHSRFFWFVTRFDIVPRITLARKASYEETLPWl 233
L P C»TFG-PLVG- I-SHAL RE W-R-F-FV -D-VPR. LA -S-E L -
Sbjct 152 LHPYCHTFG$PLVGkKIWSHALRREHWARYFLHFWKYQWPRWLAPL$$lQ€LLQVrS 211
Query
Sbjct
TGGALAAFTALWLLSQSSPPSFRVlCITFGSPLLGWQS
STSISRSRLAHHFCHWSim
S -I - R A -FCHW5 HO
GGA»A» LWLL - V CITFGSP-LGNQ5 , . _____
IGGAIASLLTLHLLCRLQTIC-SVI CITFeSfflUWQS SRAIlQKlWHFCHWSiKi
Query Sbjct Query Sbjct Query Sbjct
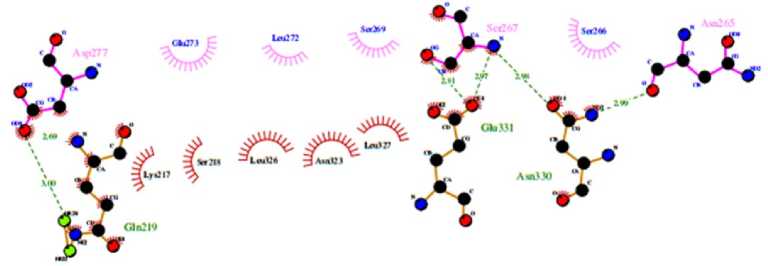
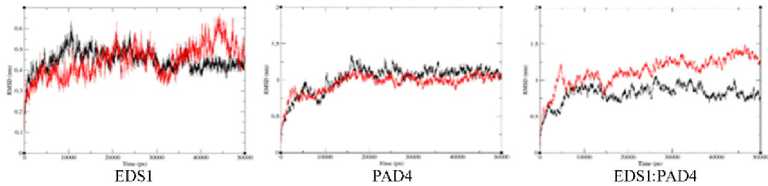
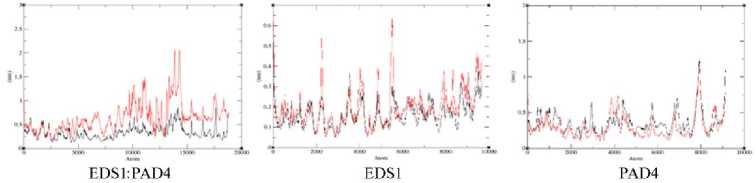
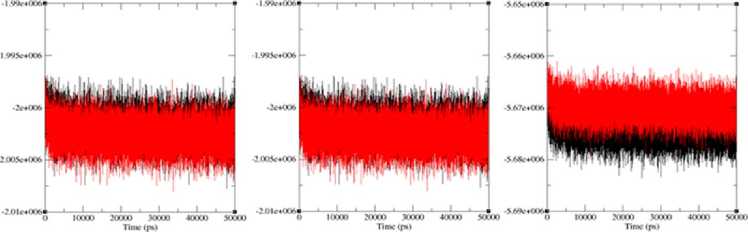
209 AQLDPRNSSVQES-—EQRITEFYTSVHREITSTVAHQAVCELTGSAEAILETLSSFLEL 264
-P- Q • F* »VHR SWA* C L 6 -LET-S- »»L
212 PFINPKSQYYQHEAVARSSHASNFFMTWRSASSVASYOACNLKGCTNLLLETVSMVQL 271
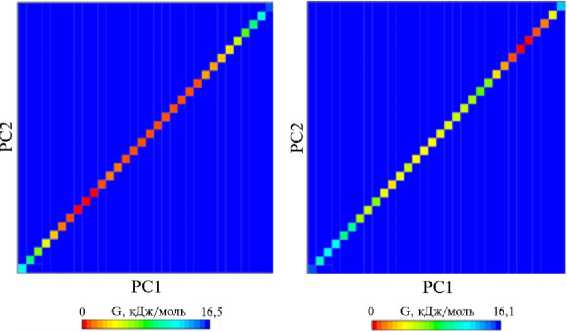
265 SPYRPAGT VrST-EKRLVAW60AlLQF'4.FYKQASDEQ€WSLlPFRSIMJrt SPYRP GT »F T «LV V N DA-LQ-LFY* Q S E E »*♦ RS» OH Y» E 272 SPYRPFGT JFCTGNRKLVWEFWAVLQLLFYSSQLSSEAEAAVVVPRSLMDHLLYKNE 331 323 LVQSHGHKLFWLOG..........ENSIESSLNDLGVSTRGRQYVQMLEEEKKRVENQ 372 • S» И» HL» • S* S»LHDLG»$TR R -AA • • ♦ 332 HQDSLEHQOVLHLNNLTOIPlSSWVOPSHNSALHDLGLSTRARLCLRAA—GErtKQKK 388 Query 373 KKHQVIQQERFLKKUMH)eYICFKQAI К - Q .R - EY* КС К GYYD*FK* N —OF AW*R ELAS** Sbjct 389 M.iEKIEQ^RSIRDALSUQEYQTKCDIRKVGVYDAFKIQ^WFI^^^ 448 Query 433 DEVLGLLKKCQLPOEFEGOIDWIKLATRYRR VEPLDIANYHRHLXHEDTGPYNKRGRPT 492 DE- +LK+ -IPO FEG DWI-L T—RR VEPLDIAKY»WLXHEDTGPY» R R? Sbjct 449 DEHEHLKRYELPKFEGRRDWIELGTQFRR VEPLDIAHYVWOIIEDTGPYURARW 533 Query 493 RYIYiQRGYEFHILKPNGHIAEOVFWNKVNGLNLGlQLEEIQETLKIISGSECGSCFKAEV 552 RY ♦ QR EH ♦ ^ •(. SCFKAEV Query 553 EELKGkPYEEVEVRVKTlEGMLREwITAGEvDEKEIFLEGSTFRKhwITLPkhhxSHS 610 EEL» КР» «V» RV LE Hi » ♦ » »IF ST» UM UP Ж $ Sbjct 538 EELRHKPFAQVQDRVLNLETAAHGWlQSSLLGD-DlFFPESTYTKhWTLPPQHKQAS 594 Query Sbjct 179 LVPR..................................SSHEQFwPFGTYLFCSDKGGVC »VPR F P G»ylFC—KG VC 189 IVPRLFFAPSCCFQFISYEHKTQLFMWLDSLGWSRGECKSSFCPSGSYLFCTIIKGAVC Query 235 LDHRGSV-RLHFHLNnATQNTEEHQRYGHYVFTLSHHFlKSRSFLGGSIPOHSYQAG ♦ON V L F -LN** * * E*H Y ♦* * F—RSF GSIP -SY-AG Sbjct 249 VDHGMWIKLLYFTLLNSSQSSSLEDHLDYADFIQKVQWQFIENR5FTEGSIPKSSYKAG Query 263 VALAVEALGFSICDT5GVLWECIETATRTVRAPILR5AELAJIELA5VLPARLEIQHYX0 * LA»E4G +♦ * • KE — A — R L SA LA L* - P R *I»K*K Sbjct 339 ITlALESlGIASHEVHFEOAKEALKKAXKLGRTRNLNSANLAIGLSKIHPFRAQIEhTKA Query 323 RCDASEEQLGYYDFFK-RySLKRDFI VWSRIRLAKFWOTVI HVETNELPFDFHIGKKW CO S EQ*GYYD FK R » KR Fl V№3»RI+LA»FWO*»I »E NELP+DFH KW Sbjct 369 SCD(6AEQMGYYDSFKQRGASKRGF)VTWIRIKLAQFWDSLI KLEAHELPYDFHKRAKW Query 382 iyasqfyqllaepldiahfykhrdiktgghylegwkryevidkwqkgvkvpee-cvrs • A$QFY»L» EPLDIA »Y» GHY»» R *RY— DKW К * RS Sbjct 429 VHASQFYKLWEPLDIAEYYRTG4ILVK6HYMQHGRERRYKIFDKkWKTENDTDHPTARS Query 441 RYASnQOTCFWAKLEQAKEWLDEARKESSDPQRRSLLREKrVPFESYANTLVTKKEVSL R-AS-TQD-CFW—E-A— L * R E D ** * E * F* YA L* KE+S Sbjct 489 RFASSTQOSCFWARVEEARDSLIKVRAE-GDARKFLKnLEOVTKFDQYAKRLIEIIKEISQ Query 501 DVKAKNSSYS 510 OV AKNSSY» Sbjct 548 DVLAJCnSSYT 557 EDSl PAD4 Рис. 1. Сравнительный анализ последовательностей EDS1 (слева) и PAD4 (справа) у Arabidopsis thaliana (образец из базы данных) и Solanum lycopersicum (исследуемый образец): синий — консервативные регионы, красный — нуклеофильный изгиб. Молекулярное моделирование. При изучении структурных особенностей EDS1 и PAD4 применяли гомологическое моделирование (38) в программе Modeller. В качестве шаблонов для EDS1 и PAD4 использовали соответственно 4NFU_A и 4NFU_B, полученные с помощью программы Blastp в NCBI. Для 4NFU_A идентичность с последовательностью EDS1 составляла 42 %, для 4NFU_B идентичность с PAD4 составляла 25 %. Оба шаблона вместе с последовательностями EDS1 и PAD4 были использованы для построения файла выравнивания. В результате было сгенерировано 10 структур. Для всех этих моделей рассчитывали значения DOPE (табл. 1). 1. Значения DOPE Scores (Discrete Optimized Protein Energy) для EDS1 и PAD4, полученные в программе Modeller v9.15 при оценке качества сгенерированных белковых структур при молекулярном моделировании Модель EDS1 PAD4 Model_1 -69354.40625 -48451.47266 Model_2 -69622.61719 -47244.53906 Model_3 -69799.39063 -47151.11719 Model_4 -69592.65625 -47395.59766 Model_5 -69583.67188 -47673.15625 Model_6 -69141.12500 -46360.92578 Model_7 -69791.44531 -46957.91016 Model_8 -69571.35156 -46751.72656 Model_9 -68983.39063 -46153.08203 Model_10 -69783.50000 -48528.74609 2. Результаты проверки полученных моделей EDS1 и PAD4 с помощью разных серверов Сервер Параметр EDS1 PAD4 PROCHECK Наиболее предпочтительная область, % 91,2 84,3 Дополнительно разрешенные области, % 7,1 13,4 Разрешенные области без ограничений, % 1,1 1,1 Запрещенные области, % 0,5 1,1 Общий G-фактор 0,01 0,08 Verify3D Средний балл 3D-1D > 0,2 79,73 65,4 ERRAT Общее качество 87,374 89,298 ProSA Z-score -10,65 -2,54 MolProbity Отклонения Св > 0,25 А, % 0,08 0,18 Остатки со слабыми связями, % 0,02 0,02 Остатки с «плохими» углами, % 0,17 0,15 VADAR Объединенное стандартное отклонение 0,13 0,71 Средняя энергия водородной связи 0,17 0,14 PROSESS Усредненное стандартное отклонение 13,45 12,59 Наибольшее стандартное отклонение (Chi score) 0,86 0,91 Спираль, % 46 50 Бета-цепь, % 10 10 Повороты, % 30 20 Витки, % 44 40 GeNMR Ошибка эффективного прогноза (Bump Score) 0,003 0,01 Радиус инерции 2,69 8,62 После перекрестной проверки параметров валидации были выбраны окончательные структуры, которые загрузили в базу данных моделей белков (PMDB), запись для EDS1 PMDB ID PM0080477, для PAD4 — ID PM0080479. PMDB — это база данных, которая собирает 3D-модели белков, построенные вручную с использованием различных методов моделирования. Для дальнейшего анализа были выбраны пять моделей с наиболее удачными показателями DOPE. Энергию систем оптимизировали для пяти выбранных моделей с использованием сервера GalaxyRefine. Проверка качества структуры. Пять отобранных моделей были дополнительно проверены с помощью сервера валидации и анализа структуры (Structure Validation and Analysis Server, SAVES). SAVES учитывает несколько параметров — карта Рамачандрана (Ramachandran Plot), число контактов низкого качества и максимальное отклонение угла/связи (табл. 2). Окончательные модели, выравненные вместе с их шаблонами, представлены на рисунке 2. EDS1 и 4NFU_A PAD4 и 4NFUB RMS = 1,461 RMS = 1,461 Рис. 2. Результаты выравнивания структур EDS1 и PAD4, сгенерированных методом молекулярного моделирования, с их шаблонами. Окончательно энергия моделей, рассчитанная в Swiss PDB Viewer, для EDS1 и PAD4 составила соответственно -26751,348 и -16361,289 КДж/моль. После минимизации энергии с помощью Swiss PDB Viewer для обоих белков значения энергии снизились до -36715,977 и -30080,314 кДж/моль. Карта Рамачандрана (Ramachandran Plot) показала, что 91,2 и 84,3 % остатков для EDS1 и PAD4 находились в коровой области, что было подтверждено сервером VADAR. Для дальнейшей оценки структур использовали сервер ProSA; Z-scores для EDS1 и PAD4 составили соответственно -10,65 и -2,54. Z-scores отражают общее качество модели; меньшее значение означает большую значимость (39). Анализ длин связей и углов проводили с помощью MolProbity, который оценивает качество модели белков как локально, так и на глобальном уровне (40). С помощью программы PROSESS средние значения Chi-1 и стандартные отклонения составили 13,45 и 12,59, то есть также находились в пределах соответствующего диапазона. Другие параметры, а также их значения приведены в таблице 2; карты Рамачандрана для обоих белков представлены на рисунке 3. Most favored region, % 91,2 Additionally allowed regions, % 7,1 Generously allowed regions, % 1.1 Most favored region, % 84,3 Additionally allowed regions, % 13,4 Generously allowed regions, % 1,1 • Core Allowed Generous Disallowed ■ Allowed * Glycine * Disallowed Рис. 3. Карты Рамачандрана (Ramachandran Plots), отображающие распределение аминокислотных остатков в различных областях, для сгенерированных методом молекулярного моделирования EDS1 (А) и PAD4 (Б). Белок- белковое взаимодействие. Финальные модели использовали в качестве исходных данных в LIGSITE для поиска остатков, входящих в активные сайты обоих белков. В активном сайте EDS1 присутствовали остатки Gly7, Ile8, Met23, His26, His124, Lys167, Ile168, Arg180, Ile214, Asn215, Pro216, Glu293, His325, Leu326, His343, Asn346 и Ile350; в активном сайте PAD4 — остатки Leu14, Val18, Leu60, Ala62, Ser129, Arg169, Val190, Ser237, Cys248, Leu272 и His275. Эти остатки были использованы в качестве исходных данных при анализе HADDOCK для изучения белок-белковых взаимодействий. HADDOCK обнаружил 131 структуру в 16 кластерах, что составляло 65,5 % для моделей с учетом водного окружения. В Discovery Studio 3.5 для дополнительного анализа были выбраны структуры с Z-score -1,6. На рисунке 4 показан финальный комплекс. Рис. 4. Финальный комплекс белков EDS1 и PAD4, сгенерированных методом молекулярного моделирования, где EDS1 представлен в виде поверхности, а PAD4 — в виде ленточной модели. При исследовании нескольких межмолекулярных взаимодействий между EDS1 (в качестве рецептора) и PAD4 (в качестве лиганда) в Discovery Studio было обнаружено, что финальный комплекс имел 13 водородных и 5 гидрофильных связей (табл. 3). С помощью LIGPLOT были выявлены различные водородные связи. Для уточнения межмолекулярных взаимодействий между EDS1 и PAD4 были построены карты контактов с указанием диапазона расстояний, связывающих оба белка (рис. 5). Определение энергии связывания белков помогает понять их взаимодействие. Связывание белков оценивают по разнице между свободной энергией двух белков: ∆G = ∆GProtein1 - ∆GProtein2. Энергия связывания для комплексов EDS1 и PAD4, согласно расчетам сервера Patchdock, составила -21,24 кДж/моль. 3. Детали взаимодействия между EDS1 и PAD4, комплекс белков EDS1 и PAD4, сгенерированных методом молекулярного моделирования ¹ структуры Обозначение Дистанция Категория Взаимодействующие структуры обозначение роль обозначение роль 1 A:ASN27:HD22-B:ASP35:OD2 2.03252 Водородная связь A:ASN27:HD22 H-Донор B:ASP35:OD2 H-Акцептор 2 A:SER218:HN-4B:ASP277:OD1 3.08046 Водородная связь A:SER218:HN H-Донор B:ASP277:OD1 H-Акцептор 3 A:GLN219:HN-B:ASP277:OD1 1.75635 Водородная связь A:GLN219:HN H-Донор B:ASP277:OD1 H-Акцептор 4 A:GLN219:HE21-B:ASP277:OD1 2.02043 Водородная связь A:GLN219:HE21 H-Донор B:ASP277:OD1 H-Акцептор 5 A:ASN330:HD22-B:ASN265:O 2.09949 Водородная связь A:ASN330:HD22 H-Донор B:ASN265:O H-Акцептор 6 A:ASN330:HD22-B:SER271:OG 2.82450 Водородная связь A:ASN330:HD22 H-Донор B:SER271:OG H-Акцептор 7 B:LYS59:HN-A:Met23:O 2.75967 Водородная связь B:LYS59:HN H-Донор A:Met23:O H-Акцептор 8 B:GLY61:HN-A:ASN20:OD1 2.65572 Водородная связь B:GLY61:HN H-Донор A:ASN20:OD1 H-Акцептор 9 B:SER267:HN-A:ASN330:OD1 2.20888 Водородная связь B:SER267:HN H-Донор A:ASN330:OD1 H-Акцептор 10 B:SER267:HN-A:GLU331:OE1 2.57991 Водородная связь B:SER267:HN H-Донор A:GLU331:OE1 H-Акцептор 11 B:SER267:HG-A:GLU331:OE1 1.83699 Водородная связь B:SER267:HG H-Донор A:GLU331:OE1 H-Акцептор 12 B:SER266:CB-A:ASN330:O 3.73266 Водородная связь B:SER266:CB H-Донор A:ASN330:O H-Акцептор 13 B:SER266:CB-A:GLU331:OE1 3.54058 Водородная связь B:SER266:CB H-Донор A:GLU331:OE1 H-Акцептор 14 A:PRO216-B:LEU276 4.93452 Гидрофобные взаимодействия A:PRO216 Алкильный радикал Alkyl B:LEU276 Алкильный радикал 15 A:LEU326-B:LEU272 5.04399 Гидрофобные взаимодействия A:LEU326 Алкильный радикал B:LEU272 Алкильный радикал 16 B:VAL38-A:LEU28 4.71876 Гидрофобные взаимодействия B:VAL38 Алкильный радикал A:LEU28 Алкильный радикал 17 B:LYS59-A:Met23 4.94864 Гидрофобные взаимодействия B:LYS59 Алкильный радикал A:Met23 Алкильный радикал 18 A:TYR220 B:ALA279 5.46945 Гидрофобные взаимодействия A:TYR220 Пи-орбитали B:ALA279 Алкильный радикал Рис. 5. LIGPLOT-карта взаимодействия EDS1 и PAD4, сгенерированных методом молекулярного моделирования. Карта отображает межмолекулярные контакты на увеличивающихся расстояниях, причем различные цвета (красный, желтый, зеленый и синий) обозначают контакты в пределах соответственно 7 А, 10 А, 13 А и 16 А. Молекулярно-динамическое моделирование и анализ. Моделирование проводили для EDS1, PAD4 и комплекса EDS1:PAD4 (GROMACS, сервер ASHOKA). Для этого оба белка и комплекс были помещены в кубическую ячейку и сольватированы H2O. Как оказалось, система EDS1 нейтральна; для стабилизации систем PAD4 и комплекса были добавлены ионы натрия (Na+) (табл. 4). 4. Число компонентов в системах EDS1 и PAD4, сгенерированных методом молекулярного моделирования, и их комплекс EDS1:PAD4 ¹ структуры Белок Число молекул H2O Число ионов Na+ Общее число атомов в белке Общее число атомов в системе 1 EDS1 48220 0 9664 57884 2 PAD4 64000 3 9136 73139 3 EDS1:PAD4 138823 4 18801 157628 После 50 нс моделирования с использованием GROMACS определяли паттерны стабильности как отдельных белков, так и комплекса. Были оценены определенные параметры с их начальными и конечными значениями. RMSD показывает изменения в конфигурации белка во время моделирования. Большие изменения RMSD означают, что белок претерпел большее конформационное изменение, и наоборот (41). Через 50 нс средние значения RMSD для EDS1, PAD4 и EDS1:PAD4 (по два запуска для каждого при одинаковых условиях) составили соответственно приблизительно 4,3; 10,1 и 1,2 А. Для PAD4 значение RMSD было очень большим, что позволяет предположить возможность значительных конформационных изменений во время моделирования. Значение RMSD для EDS1:PAD4 не имело столь существенного отклонения (рис. 6). Рис. 6. Графики RMSD для белков EDS1, PAD4 и их комплекса при молекулярно-динамическом моделировании (50 нс). RMSF рассчитывали для определения аберрации между положениями атомов. RMSF отражает изменение положения для каждого остатка в белке (42). Среднее значение RMSF всех атомов для EDS1, PAD4 и их комплекса составило соответственно 0,38; 0,94 и 0,70 нм. Максимальное отклонение позиций атомов наблюдалось в EDS1:PAD4; между EDS1 и PAD4 большее отклонение во время моделирования показал PAD4 (рис. 7). Рис. 7. Графики RMSF для белков EDS1, PAD4 и их комплекса при молекулярно-динамическом моделировании (50 нс). Еще один важный параметр — потенциальная энергия. Ее изменение рассчитывали в интервале времени от t = 0 нс до t = 50 нс. Для EDS1 потенциальная энергия при t = 0 нс составила -2000427,00 кДж/моль, для PAD4 — -2607008,00 кДж/моль, для EDS1:PAD4 — -5667363,00 кДж/моль. К t = 50 нс потенциальная энергия изменилась соответственно до -2002373,00, -2614066,38 и -5672486,50 кДж/моль для EDS1, PAD4 и EDS1:PAD4 (рис. 8). Для всех трех белков не наблюдалось значительных изменений потенциальной энергии. Анализ методом молекулярной динамики показал, что могли произойти некоторые конформационные изменения, особенно в PAD4, но компактность PAD4 увеличилась по сравнению с EDS1. EDS1 PAD4 EDS1:PAD4 Рис. 8. Графики потенциальной энергии для белков EDS1, PAD4 и их комплекса при молекулярно-динамическом моделировании (50 нс). Площадь доступной для растворителя поверхности (solvent-accessible surface area, SASA) также была рассчитана примерно для 50 нс для обоих белков и их комплекса. Для EDS1 общая SASA изменилась с 325,592 нм2 (площадь гидрофобной поверхности — 106,769 нм2, площадь гидрофильной поверхности — 218,823 нм2) в момент времени t = 0 нс до 335,951 нм2 (площадь гидрофобной поверхности — 108,543 нм2, площадь гидрофильной поверхности — 227,407 нм2) при t = 50 нс. Для PAD4 общая SASA изменилась с 389,8 нм2 (площадь гидрофобной поверхности — 143,343 нм2, площадь гидрофильной поверхности — 246,457 нм2) при t = 0 нс до 396,861 нм2 (площадь гидрофобной поверхности — 144,94 нм2, площадь гидрофильной поверхности — 251,921 нм2) при t = 50 нс. SASA EDS1:PAD4 изменилась с 718,038 нм2 (площадь гидрофобной поверхности — 252,663 нм2, площадь гидрофильной поверхности — 465,375 нм2) при t = 0 нс до 727,793 нм2 (площадь гидрофобной поверхности — 247,483 нм2, площадь гидрофильной поверхности — 480,31 нм2) при t = 50 нс. Общая площадь поверхности взаимодействия была дополнительно рассчитана с использованием уравнения [1]. Взаимодействие EDS1 и PAD4 происходило на площади 5,019 нм2, что привело к образованию 18 связей. Из них 13 были водородными, 5 — гидрофобными (см. табл. 4). В EDS1 и PAD4 в белок-белковом взаимодействии участвовали соответственно 11 и 12 аминокислотных остатков. Анализ основных мод движения. Поскольку структурные перестройки важны для правильной передачи сигналов (43) и общего функционирования, движение EDS1 и PAD4 было сопоставлено с движением белка посредством расчетов основных мод. Для изучения движения остатков проанализировали динамическую кросс-корреляцию. Значение корреляции составляло от -0,0913 (синий) до 0,271 (красный) для EDS1 и от -0,505 (синий) до 1,6 (красный) для PAD4, что представляет собой соответственно отрицательную и положительную корреляцию. На рисунке 9 представлена матрица, соответствующая ковариации между всеми атомами Cα. Красный цвет означает, что два атома движутся вместе и, следовательно, лежат по диагонали, тогда как синий показывает движение атомов в противоположных направлениях. Как видно, флуктуаций было меньше. Рис. 9. Анализ главных компонент (Principal Component Analysis, PCA) для EDS1 и PAD4, сгенерированных методом молекулярного моделирования. Липазные домены в EDS1 находились в позиции от 46-го до 200-го аминокислотного остатка, в PAD4 — от 92-го до 196-го остатка. Корреляция между атомами в этих доменах была высокой. Помимо этого, при анализе PCA также учитывалась свободная энергия Гиббса (рис. 10). Была обнаружена тесная взаимосвязь между структурой и функцией белка, опосредованная энергетикой. Была выявлена статистическая корреляция между ∆G и структурными изменениями белка (44). Результаты анализа белок-белковых взаимодействий и молекулярной динамики подтвердили существование комплекса EDS1 и PAD4, что, в свою очередь, указывает на проявление салицилатного сигнального пути у томата. При участии R-генов инфицирование растений приводит к активации синтеза салициловой кислоты. Для должного накопления SA необходимы EDS1 и PAD4 (45). Анализ последовательностей выявил домены семейства липаз класса 3, ответственные за различные реакции растений на стресс (46). Присутствие этих доменов в EDS1 и PAD4 подразумевает, что оба белка участвуют в реакциях растений на стресс. Структурный анализ касался взаимодействия двух белков. Было отмечено, что PAD4 претерпевает более значительные конформационные изменения, чем EDS1. Конформационные изменения также наблюдались в комплексе EDS1:PAD4 в течение 50 нс моделирования. Между EDS1 и PAD4 было также выявлено значительное число водородных связей и гидрофобных взаимодействий. Анализ главных компонентов показал, что атомы в липазном домене перемещались согласованно как в EDS1, так и в PAD4. Вышеизложенные наблюдения позволяют предположить, что белки EDS1 и PAD4 могут взаимодействовать в растениях томата, хотя этот вывод требует дальнейшего экспериментального подтверждения. EDS1 PAD4 Рис. 10. Графики свободной энергии Гиббса для EDS1 и PAD4, сгенерированных методом молекулярного моделирования. Таким образом, в процессе динамических биоимитационных исследований была успешно смоделирована ранее не описанная трехмерная структура белков EDS1 и PAD4, что позволило выявить существование молекулярного взаимодействия между EDS1 и PAD4 за 50 нс и подтвердить стабильность структуры их комплекса. Параметры моделирования молекулярной динамики (МД), среднеквадратичное отклонение (RMSD), среднеквадратичные флуктуаций (RMSF), площадь доступной поверхности (SASA), потенциальная энергия и анализ главных компонентов (PCA) подтвердили стабильность комплекса. Докинг-анализ выявил наличие 13 водородных связей и 5 гидрофобных взаимодействий между этими белками. Все эти данные указывают на то, что салициловый путь может функционировать в томатах в условиях биотического стресса. В будущих исследованиях следует идентифицировать ключевые гены, связанные с салициловым путем, и их маркеры, которые могут быть использованы в молекулярной селекции для программ улучшения генетического материала. Мы выражаем глубокую благодарность Индийскому совету сельскохозяйственных исследований (ICAR) при правительстве Индии за поддержку в создании Центра передовых суперкомпьютерных вычислений для получения омиксных знаний в сельском хозяйстве (ASHOKA) в ICAR-IASRI, Нью-Дели. Частично работа была поддержана программой CABin Scheme (код проекта: 1004936) ICAR-IASRI, Нью-Дели. Мы также высоко ценим поддержку и конструктивную критику, полученные от заведующего кафедрой вычислительной биологии и биоинформатики и декана Института биотехнологии и биоинженерии им. Джейкоба при Университете сельского хозяйства, технологий и наук им. Сэма Хиггинботтома, Ирайаградж. 1Division of Agricultural Bioinformatics, ICAR-Indian Agricultural Statistics Research Institute, New Delhi-11012, India,