Молекулярное моделирование распознавания стоп-кодона фактором терминации трансляции 1-го типа ERF1

Автор: Мазур Ю.А., Опарина Н.Ю.
Журнал: Труды Московского физико-технического института @trudy-mipt
Рубрика: Оригинальные статьи
Статья в выпуске: 1 т.1, 2009 года.
Бесплатный доступ
Короткий адрес: https://sciup.org/142185570
IDR: 142185570
Текст статьи Молекулярное моделирование распознавания стоп-кодона фактором терминации трансляции 1-го типа ERF1
Терминация трансляции — важнейший завершающий этап синтеза белка, при котором обеспечивается высокая точность распознавания «бессмысленных» стоп-кодонов и новосинтезирован-ные полипептиды диссоциируют от рибосом. В отличие от прокариот, у которых декодирование трёх стоп-кодонов UAA, UAG и UGA осуществляется с помощью двух паралогичных факторов терминации RF1 и RF2, отличающихся специфичностью по отношению к стоп-кодонам, у эукариот и архебактерий все три стоп-кодона с одинаковой эффективностью декодируются одним белковым фактором — eRF1/aRF1 [1– 4]. Собственно распознавание стоп-кодонов происходит после связывания фактора eRF1/aRF1 с A-сайтом рибосомы, однако есть ряд свидетельств, что основную роль в специфическом декодировании играет не рибосома, а eRF1/aRF1, точнее его N-концевой домен [5–10]. Применение методов случайного [11] и направленного мутагенеза [8, 9] позволили выявить в N-концевом домене eRF1 ряд аминокислотных остатков, замены в которых меняют специфичность декодирования стоп-кодонов. Однако несмотря на расположение в одном домене и сближенность некоторых из выявленных участков, в целом они расположены в различных областях декодирующего домена eRF1 и не образуют четких пространственных кластеров. Одним из многообещающих подходов для выявления механизма распознавания стоп-кодона служит исследование филогенетических групп, в которых наблюдают отклонения от стандартного генетического кода. В частности, для инфузорий Stylonychia, Tetrahymena и Paramecium стоп-кодоном служит только UGA, а триплеты UAA и UAG кодируют глутамин, а для Euplotes показано использование в качестве стоп-кодонов только UAA и UAG, но не UGA, который кодирует цистеин [7, 12–15]. В перечисленных работах были предприняты попытки выявления области eRF1, отвечающей за специфическое декодирование стоп-кодонов, с помощью сравнительного анализа выравненных аминокислотных последовательностей eRF1, различающихся набором распознаваемых стоп-кодонов. Однако и в этом случае не выявили четкого кластера аминокислотных замен, который можно было бы считать потенциальным сайтом декодирования. На основании сравнения eRF1 из групп с одинаковым изменением специфичности распознавания стоп-кодонов экспериментально показано, что, по-видимо-му, один и тот же тип изменения специфичности связывания N-концевого домена eRF1 со стоп-кодонами можно создать рядом независимых способов с использованием пространственно разнесенных аминокислотных замен [10, 14].
Следует отметить, что во всех перечисленных случаях имеющиеся экспериментальные данные позволяют сопоставлять способность eRF1 распознавать только 2-й и 3-й пуриновые нуклеотиды стоп-кодона (AA, AG или GA) и отличать их от 2-го и 3-го гуаниновых нуклеотидов триптофанового кодона UGG, сведения же о распознавании первого инвариантного урацила стоп-кодонов незначительны.
В нашей работе проведено молекулярное моделирование образования комплексов N-концевого домена eRF1 человека (PDB 1dt9) и «специфических» рибодинуклеотидов AA, AG и GA в сравнении с «неспецифическим» рибодинуклеотидом GG.
-
II. Материалы и методы
-
II.1. Исходные данные
-
Для анализа взаимодействия N-концевого домена белка eRF1 человека с пуриновыми динуклеотидами стоп-кодонов и родственным «неспецифическим» динуклеотидом GG использована структура 1DT9 из банка данных RCSB (. Из структуры полноразмерного белка eRF1 была вырезана с использованием пакета программ Sybyl ( структура N-концевого домена, состоящая из аминокислотных остатков 5-140. С помощью программы Biopolymer из пакета программ Sybyl были получены модели рибодинуклеотидов PO4-5’-AA-3’-OH, PO4-5’-AG-3’-OH, PO4-5’-GA-3’-OH и PO4-5’-GG-3’-OH в виде одноцепочечных фрагментов A-формы
РНК. Необходимые точечные мутации в комплексы N-домен+dNTP вносились с помощью программ SwissPDBViever и пакета программ Sybyl.
-
II.2. Молекулярный докинг
Молекулярный докинг пуриновых рибонуклеотидов на N-концевой домен eRF1 человека выполнен с помощью программы Autodock 3.0 в отсутствие начальных ограничений. Для оптимизации скоринг-функции применяли параметры для докинга РНК (Moitessier et. al., 2006) [16]. Использован генетический алгоритм для автоматического докинга. Для каждого рибодинуклеотида проводили 100 независимых раундов молекулярного докинга.
-
II.3. Анализ результатов молекулярного докинга и кластеризация потенциальных моделей комплексов N-концевого домена eRF1 с рибодинуклеотидами
Среди вариантов расположения лигандов на первом этапе были выбраны только те, для которых энергия взаимодействия согласно скоринг-функции программы Autodock была минимальна.
Далее с помощью in-house программы фильтрации результатов молекулярного докинга были рассчитаны межмолекулярные взаимодействия для РНК-белковых комплексов (включая водородные связи и ван-дер-ваальсовы взаимодействия) и применены следующие фильтры: отсутствие плотного (ближе 4Е) контакта с белком для 5’/3’ концов рибодинуклеотида (данный параметр определяется тем, что изучаемые рибодинуклеотиды на деле представляют собой фрагменты мРНК); основная часть (более 80% ) межмолекулярных взаимодействий для рибодинуклеотида должна приходиться на азотистые основания, а не на сахарофосфатный остов; наличие среди контактов не менее 3 связей с длиной до 3,5Е; соотношение контактов между первым и вторым пуринами не должно превышать 3:1 (этот параметр введён, чтобы избежать отбора стабильных комплексов, для которых большая часть РНК-белковых взаимодействий приходится только на один из нуклеотидов, что не позволяет считать такие взаимодействия специфичными).
На втором этапе все модели комплексов, прошедшие фильтр, были классифицированы согласно их пространственному расположению на N-концевом домене eRF1 (согласно совпадению аминокислотных остатков, взаимодействующих с 1-м и 2-м пуринами рибодинуклеотида соответственно). По результатам работы программы были построены кластеры возможных специфичных РНК-белковых взаимодействий с использованием пакета программ CLUTO (glaros.dtc.umn.edu/gkhome/views/cluto) и in-house разработанного программного продукта. Для каждого из кластеров были посчитаны средние энергии, и если эти средние энергии оказывались отрицательными, то из таких кластеров отбиралась лучшая модель и далее использовалась для молекулярной динамики.
-
II.4. Молекулярная динамика
Молекулярная динамика отобранных в докинге моделей была выполнена с помощью пакета программ Amber. Были использованы условия непрямого указания растворителя. Последовательность действий при молекулярной динамике была следующей: этап минимизации структуры, медленный нагрев от 0 до 300 Ки проведения динамики при 300 К до достижения равновесного состояния. Поскольку параметры диэлектрической проницаемости рибосомы в области A -сайта неизвестны, была выбрана условная единица 20, основанная на большей гидрофильности РНК, нежели внутренняя часть белковой глобулы (где диэлектрическая проницаемость равна 4). Аналогичная процедура проводилась для свободного N-домена eRF1 и для свободных динуклеотидов AA, AG, GA и GG. Это позволило определить потенциальную энергию в равновесном состоянии и разницу этой энергии для комплекса и для пары «свободный белок + свободный лиганд».
Кроме того, для комплексов AG/eRF1 и GA/eRF1 получены «мутантные» структуры, в которых искусственно аденин был заменен на гуанин (программа SwissPDBViewer), при этом расположение нуклеотидов не меняли. Молекулярная динамика этих комплексов была проведена при тех же условиях.
-
III. Результаты
При проведении «слепого» молекулярного докинга подавляющее большинство моделей соответствовало расположению лиганда в области, близкой к функционально важному Y-C-F району [1, 8–9]для каждого из динуклеотидов AA, AG, GA и GG. Однако отобранные модели не позволяли с помощью фильтрации по энергии взаимодействия, определённой с помощью скоринг-функции Autodock 3.0, выявить более локальный участок взаимодействия. Поэтому нами была применена система фильтрации моделей комплексов, необходимая для отбора потенциально специфических комплексов динуклеотид — eRF1 (см. материалы и методы). При этом были отобраны кластеры моделей расположения пуринового динуклеотида в комплексе с eRF1 (рис. 1). Однако на основании одного только сравнения энергий взаимодействия также не удаётся определить сайт декодирования стоп-кодона в eRF1. Нами было замечено, что среди выявленных ориентаций динуклеотидов AA, AG и GA присутствует чрезвычайно близкое направление (отмеченное черной стрелкой), в то же время комплексов с аналогичным расположением динуклеотидного лиганда для моделей связывания GG и eRF1 не обнаружено (существует лишь близкое направление). Это позволило нам предположить, что в отличие от РНК-белковых комплексов, образующихся в растворе, специфический комплекс eRF1–стоп-кодон не способен образовываться в отсутствие рибосомы, которая обеспечивает правильное взаимное расположение домена 1 eRF1 и стоп-кодона в мРНК. Таким образом, модели комплексов динуклеотидов AA, AG и GA, соответствующие общему кластеру расположения лиганда, отсутствующему среди комплексов eRF1 с GG, были отобраны для дальнейшего исследования с помощью молекулярной динамики. Как указывалось выше (см. материалы и методы), для дальнейшего анализа также были использованы представители других кластеров.
Для более точного анализа взаимодействий в комплексе РНК–белок, а также для оценки его стабильности была проведена молекулярная динамика отобранных моделей. На основании анализа энер- гий взаимодействия домена 1 eRF1 со специфическими динуклеотидами AA, AG и GA и с неспецифическим GG были отобраны три модели (модели AA_82, AG_66, GA_96), по 1 модели для каждого специфического динулеотида, отвечающие следующим требованиям: энергии взаимодействия eRF1 со специфическими лигандами должны быть близки, но в то же время быть значительно ниже, чем энергии связывания eRF1 с неспецифическим лигандом GG (табл. 1).
Проведённый анализ контактных аминокислотных остатков показал, что аминокислоты T122, S123, L124 и Y125 близко контактируют со всеми специфическими динуклеотидами (табл. 2). Значительная часть контактных аминокислотных остатков консервативна или инвариантна для eRF1 с неизмененной специфичностью распознавания стоп-кодона.
Мы провели анализ расположения вероятных водородных связей и ван-дер-ваальсовых взаимодействий для отобранных моделей комплексов. Как видно из таблицы 3, часть определённых контактных остатков участвует во взаимодействии одновременно и с первым, и со вторым нуклеотидами лиганда (соответствуют второму и третьему нуклеотидам стоп-кодона). Однако в целом можно говорить о предпочтительном участии аминокислотных остатков 122, 123в распознавании первого, а 125 — второго нуклеотидов лиганда.

Рис. 1. Расположение кластеров динуклеотид-ных лигандов в комплексе с доменом 1 eRF1. Стрелками схематически показаны направления, характерные для кластеров. Направление стрелок указывает направление от 5’ конца мРНК к 3’ концу
Таблица 1
Энергии взаимодействия специфических и неспецифического динуклеотидов с доменом 1 eRF1
|
Потенциальная энергия |
Разница в энергии комплекса и пары (свободный белок+свободный лиганд) |
||
|
AA |
|||
|
AA_26 |
-3048,1199 |
-13,397 |
|
|
AA_82 |
-3066,478 |
-31,7551 |
|
|
AA_69 |
-3028,9956 |
5,7273 |
|
|
AG |
Сравнение с «мутантом» AG-GG |
||
|
AG_41 |
-3085,8551 |
-16,5292 |
14,7129 |
|
AG_66 |
-3094,7455 |
-25,4196 |
-2,1839 |
|
GA |
Сравнение с «мутантом» GA-GG |
||
|
GA_29 |
-3049,7892 |
21,4498 |
-26,8293 |
|
GA_34 |
-3099,3908 |
-28,1518 |
-28,8103 |
|
GA_96 |
-3103,9689 |
-32,7299 |
-6,3395 |
Таблица 2
Контактирующие с динуклеотидным лигандом аминокислотные остатки в домене 1 eRF1
|
Intersections |
Variation among eRF1s |
||
|
AA |
AG |
GA |
|
|
I38 |
Variable |
||
|
P40 |
P40 |
Conserved |
|
|
R47 |
R47 |
Conserved |
|
|
V48 |
Variable |
||
|
M51 |
Variable |
||
|
D43 |
Conserved |
||
|
P119 |
Invariant |
||
|
N121 |
N121 |
Invariant |
|
|
T122 |
T122 |
T122 |
Conserved |
|
S123 |
S123 |
S123 |
Conserved |
|
L124 |
L124 |
L124 |
Conserved |
|
Y125 |
Y125 |
Y125 |
Invariant |
|
L126 |
Conserved |
||
|
H132 |
Conserved |
||
|
E134 |
Conserved |
||
|
A135 |
A135 |
Conserved |
|
|
L136 |
Invariant |
||
|
A138 |
Variable |
||
|
L139 |
Invariant |
||
Показаны близкие ( < 3 A ) контакты для моделей комплексов с AA, AG и GA. Аминокислоты и их нумерация соответствуют eRF1 человека. Отмечена консервативность соответствующих остатков для eRF1, распознающих все три стоп-кодона. Выделены аминокислоты, контактирующие со всеми вариантами динуклеотидных лигандов.
Рахица а энергии коажэпекса и пары свободный бепокесвободныи лиганд при Е=?0
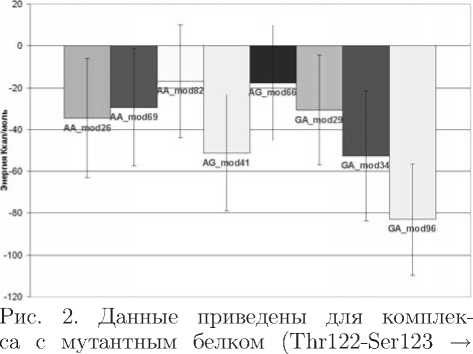
Gln122-Phe123)
В конце мы провели виртуальный мутагенез, при котором заменили две аминокислоты Thr122-Ser123 на Gln122-Phe123. После проведённой 15 пс молекулярной динамики были определены энергии взаимодействия (рис. 2). Как легко заметить, наиболее стабильным среди проанализированных вариантов оказался комплекс с GA, соответствующий стоп-кодону UGA, но не комплексы с AA и AG, представляющими фрагменты стоп-кодонов UAA и UAG. Таким образом, полученные данные полностью соответствуют известным результатам эксперимента, что подтверждает высокую специфичность взаимодействия полученных нами молекулярных моделей [10]. Необходимо отметить, что наилучшее соответствие с экспериментом показала тройка моделей AA_mod82, AG_mod66, GA_mod96, которые соответствуют немутированным моделям AA_82, AG_66, GA_96. Поскольку отбор этих моделей был независимым в двух виртуальных экспериментах, можно говорить о данных моделях как о потенциальных кандидатах в качестве специфических комплек- сов посадки мРНК на N-концевой домен eRF1 человека.
Таблица 3
Аминокислотные остатки домена 1 eRF1, образующие вероятные водородные связи и/или ван-дер-ваальсовы взаимодействия с первым и вторым нуклеотидами специфического динуклеотидного лиганда
|
Amino acid |
AA |
AG |
GA |
|
ILE38 |
A2 |
||
|
ARG47 |
A1 |
G1 |
|
|
VAL48 |
G1 |
||
|
MET51 |
G1 |
||
|
ASN121 |
A1 |
A2 |
|
|
THR122 |
A1, A2 |
A1 |
G1 |
|
SER123 |
A1 |
A1 |
G1 |
|
LEU124 |
A2 |
G2 |
A2 |
|
TYR125 |
A1, A2 |
G2 |
A2, G1 |
|
HIS132 |
G2 |
||
|
GLU134 |
G2 |
||
|
ALA135 |
A2 |
-
IV. Выводы
Впервые применены методы молекулярного моделирования для определения декодирующего участка на N-концевом домене eRF1 человека. Показано, что как для специфических пуринов стоп-кодонов (AA, AG и GA), так и для неспецифического динуклеотида GG удаётся выявить сайты связывания, близкие по пространственному расположению и энергии. Тем не менее, благодаря использованным в настоящей работе параметрам фильтрации моделей РНК-белковых комплексов удаётся выявить такое расположение пуриновых динуклеотидов на N-концевом домене eRF1, которое отвечает требованиям высокоспецифического связывания. Дополнительно проведённый виртуальный мутагенез части контактных аминокислот показал совпадение результатов молекулярного моделирования с известными ранее экспериментальными данными.
