Монте-Карло моделирование активности углерода в ОЦК-железе

Автор: Ридный Ярослав Максимович, Мирзоев Александр Аминулаевич, Мирзаев Джалал Аминулович
Журнал: Вестник Южно-Уральского государственного университета. Серия: Металлургия @vestnik-susu-metallurgy
Рубрика: Физическая химия и физика металлургических систем
Статья в выпуске: 4 т.18, 2018 года.
Бесплатный доступ
Задача исследования заключалась в проверке предложенных в литературе значений энергии взаимодействия между атомами углерода в ОЦК-железе для различных координационных сфер. Цель работы: рассчитать активность углерода в ОЦК-железе, используя различные параметры взаимодействий между атомами углерода. Методы: для моделирования использовалась самописная программа по Монте-Карло, в которой была рассмотрена суперячейка 20×20×20 элементарных ячеек объёмно-центрированного (ОЦК) железа. Нами была проведена оптимизация, в ходе которой была получена сходимость результата усреднения при 500 шагах Монте-Карло. Расчеты проводили для широкого диапазона температур (Т = 955, 975, 1000, 1026, 1056, 1070, 1086, 1121 К). Результаты: наши расчеты показали, что почти все предложенные параметризации согласуются с экспериментальными данными, поскольку при низких концентрациях атомы углерода практически не взаимодействуют. Чтобы изучить влияние взаимодействия атомов углерода на кривую активности, нам пришлось расширить наши расчеты до области более высоких концентраций, чем наблюдаются на самом деле. Это позволило нам сравнить различные параметризации и получить ряд интересных выводов. Кроме того, мы продемонстрировали возможность воспроизведения экспериментальных кривых активности углерода для нескольких наборов параметров С-С взаимодействия. Новизна работы: мы продемонстрировали, что для расчета активности углерода в ОЦК-железе достаточно вычислить энергии С-С взаимодействия для первых четырех координационных сфер. Практическая значимость заключается в получении новой теоретической информации, необходимой для разработки новых марок сталей и режимов термической обработки их.
Первопринципное моделирование, оцк-железо, активность углерода, монте-карло метод
Короткий адрес: https://sciup.org/147232513
IDR: 147232513 | УДК: 669.112.227.1:538.915 | DOI: 10.14529/met180403
Monte Carlo simulation of carbon activity in BCC iron
The research problem is to verify the literature-proposed values of carbon atomic interactions in BCC iron for various coordination spheres. The goal hereof is to compute the carbon activity in BCC iron using various carbon-atom interaction parameters. Methods: An author-coded Monte Carlo program is used for simulation; the program analyzes a supercell comprised of 20×20×20 unit cells of body-centered cubic (BCC) iron. The paper presents a successfully optimized solution that attains convergence of averaging at 500 Monte Carlo steps. Computations are performed for a wide temperature range (T = 955, 975, 1000, 1026, 1056, 1070, 1086, 1121 K). Results: Computations show that virtually all of the proposed parameterizations are consistent with the experimental data, as carbon-atom barely interact at low concentrations. Computations are further expanded to cover higher-than-real concentrations to study how carbon atoms could affect the activity curve. This enables a comparison of various parameterizations, bringing a few interesting findings. Besides, the research demonstrates that experimental carbon activity curves can be reproduced for multiple C-C interaction configurations. The novelty of this research is that computing the energy of C-C interaction for the first four coordination spheres suffices for computing the carbon activity in BCC iron. The practical significance is that the research has produced new theoretical data that will be made use of when developing new steel grades and designing new thermal-treatment processes.
Текст научной статьи Монте-Карло моделирование активности углерода в ОЦК-железе
Сплавы на основе железа, несмотря на рост производства пластмасс, керамики и композитов, продолжают оставаться основными конструкционными материалами. Это связано с тем, что с помощью относительно несложной термомеханической обработки могут быть получены метастабильная ОЦК-фаза, перенасыщенная атомами С, обладающая высокой прочностью, пластичностью и рядом важных для практики физических свойств (электро- и теплопроводность, жаростойкость, магнитная проницаемость и т. п.). Одним из основных факторов, определяющих эти свойства, является параметр взаимодействия между атомами углерода. К сожалению, энергии взаимодействия атомов растворенного вещества не могут быть непосредственно измерены в эксперименте. Основным источником информации о взаимодействии были термодинамические данные о растворах углерода в железе [1].
Оценки энергии взаимодействия атомов углерода в ОЦК-железе были получены в [2–6].
Подробный обзор взаимодействий между атомами углерода в ОЦК-железе приведен в [6]. Из-за низкой растворимости углерода в α-железе (менее 0,02 ат. %), на практике у нас будет почти бесконечно разбавленный раствор, где взаимодействия C–C не появляются. Прямой расчет зависимости энергии взаимодействия С–С от активности углерода не является продуктивным. Авторы первых работ [7, 8], где были сделаны такие расчеты, получили отрицательную энергию взаимодействия С–С. Однако они использовали неправильное координационное число для междоузельных подрешеток [9]. Позднее Бхадешиа сделал более точные расчеты [10]. Он определил энергию взаимодействия ближайших соседей углерода в железе (ECC = 1,54 эВ), используя квантовохимическую теорию активности. Это значение согласуется с энергией ECC = 1,78 эВ, полученной в [6]. В работах [2–5] получены очень различные значения энергии С–С взаимодействия в ОЦК-железе с использованием методов первопринципного компьютерного моделирования и псевдопотенциального прибли- жения для решетки железа. К сожалению, до сих пор неясно, какие данные верны.
В настоящей работе мы проанализировали текущую ситуацию и решили проблему, вычислив активность углерода в ОЦК-железе. Для расчётов использовался метод Монте-Карло с доступным набором энергий С–С взаимодействия. Для этого нам необходимо было ответить на следующие вопросы:
-
1. Почему данные по активности углерода воспроизводятся с различными наборами параметров С–С взаимодействия?
-
2. Какое из предложенных значений параметров С–С взаимодействия следует рассматривать как наиболее подходящее для расчета термодинамических свойств ОЦК-железа?
Монте-Карло метод
Для моделирования активности углерода в железе мы использовали метод, предложенный в [11] и примененный для расчета углеродной активности в железе [12]. Расчет углеродной активности в железе проводили по следующей формуле:
a С = a conf exp ( A GjkT ) , (1) где a С – активность углерода в железе; a conf – конфигурационная часть активности и неконфигурационная часть. Для вычисления неконфигурационной части использовались данные работы [13].
Идея метода заключается в следующем. Химический потенциал системы представляется через отношение статистических сумм для двух значений числа частиц – N и N + 1. Удается свести данное отношение к усредненному по ансамблю ^e вАЛ' ^, где в = 1/kT, k – константа Больцмана, T – абсолютная температура, ΔEi – изменение энергии системы из N частиц при введении виртуального атома углерода в узел i при фиксированной позиции других атомов. Усреднение выполняется по обычной процедуре Монте-Карло, в результате чего приходим к следующему выра- жению для конфигурационной части активности [14, 15]:
a conf = M ( N C + 1 )
/ Г M Г m Vi z Z exp ( / I j = 1 L i = 1
A EilkT ) ^ . (2)
Суммирование по i выполняется по всем пустым октаэдрическим междоузлиям m =3 N Fe – N C и по числу шагов j ( j =1, 2, …, M ), используемых в расчете методом Монте-Карло, N Fe и N C – число атомов железа и углерода в решетке. Для моделирования использовали систему 20×20×20 элементарных ячеек ОЦК-решётки железа. Сходимость результата усреднения достигали при числе шагов Монте-Карло порядка 500.
Вычисление активности углеродав ОЦК-железе
Как упоминалось выше, энергии C–C взаимодействия были получены с использованием первопринципного моделирования [3–6] (см. таблицу). Авторы [3–5] использовали су-перячейку из 128 атомов ОЦК-железа. В наших расчетах мы рассматривали суперячейку из 54 атомов (рис. 1). Расчеты проводили для широкого диапазона температур ( Т = 955, 975, 1000, 1026, 1056, 1070, 1086, 1121 К). Однако ниже мы представили только данные для температуры 1000 К, поскольку не наблюдалось каких-либо качественных различий в результатах. Растворимость углерода в ОЦК-железе составляет 0,1 ат. %. Мы вычислили зависимость активности углерода в ОЦК-железе от концентрации, используя данные из таблицы, и сравнили полученные кривые (рис. 2).
Все рассмотренные исследования отлично согласуются с экспериментом [16] (рис. 2а), поскольку растворимость углерода в ОЦК-железе мала. Так как атомы С расположены на достаточном расстоянии друг от друга, энергия взаимодействия С–С мала и конфигурационная часть активности в уравнении (1) близка к 1. Чтобы сравнить различные параметры взаимодействия C–C, мы экстраполи-
Энергии взаимодействия атомов углерода в ОЦК-железе, эВ
|
Координационная сфера |
1 |
2 |
3 |
4a |
4b |
5 |
6 |
7a |
7b |
8a |
11 |
|
Домэйн [3] |
– |
0,65 |
0,09 |
0,09 |
1,67 |
–0,13 |
– |
– |
–0,14 |
– |
–0,16 |
|
Бароух [4] |
1,91 |
0,82 |
0,23 |
0,02 |
1,52 |
0,06 |
0,07 |
0,02 |
0,11 |
0,02 |
– |
|
Рубан [5] |
1,95 |
0,96 |
0,34 |
0,25 |
1,9 |
–0,07 |
–0,02 |
–0,03 |
0,1 |
0,05 |
0,02 |
|
Данная работа [6] |
1,78 |
0,66 |
0,12 |
–0,13 |
2,89 |
–0,14 |
–0,1 |
–0,08 |
0 |
0,03 |
–0,19 |
Физическая химия и физика металлургических систем
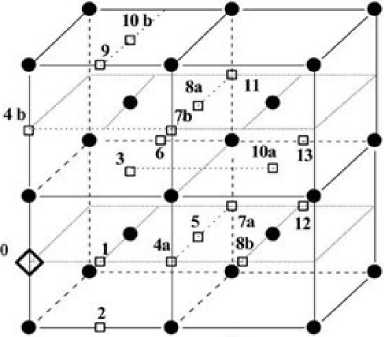
Рис. 1. Возможные положения атомов углерода в ОЦК-железе. Черные круги обозначают атомы железа; квадраты – атомы углерода. Начальный атом находится в положении 0
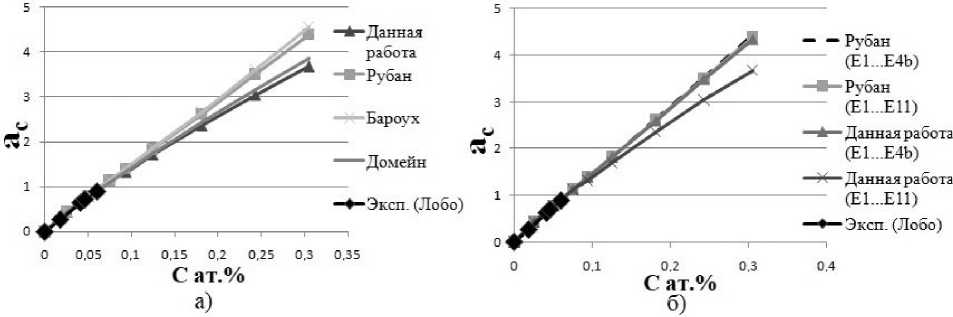
Рис. 2. Зависимость активности углерода в ОЦК-железе от концентрации. Кривые были получены методом Монте-Карло с использованием данных, приведенных в таблице
ровали кривую активности углерода в более широкий диапазон концентраций (не достижимых в реальном ОЦК-железе). После этого мы видим, что рассматриваемые исследования приводят к различным кривым активности (см. рис. 1). Кривые, рассчитанные из [3] и наших данных [6], почти совпадают, а другие две кривые (из [4] и [5]) лежат выше. Согласие наших результатов с Рубаном [3] можно объяснить тем, что он не учитывал все возможные расположения атомов углерода. Мы моделировали суперячейку из 54 атомов, в то время как в [4, 5] число атомов в супер-ячейке было в два раза больше. Это могло бы означать, что на энергии взаимодействия атомов углерода, расположенных в 5–11 координационных оболочках, влияют периодические граничные условия. Мы хотели проверить эту гипотезу и выяснить необходимость рассмотрения таких далеких пар C–C для расчета углеродной активности. Мы провели численные эксперименты с учетом взаимодействия атомов углерода, расположенных только в первых четырех координатных оболочках. Результаты подтвердили, что, не учитывая неточных значений энергий взаимодействия С–С, мы можем получить кривую активности, которая совпадает с одной, рассчитанной из более точных исследований [5] (рис. 2б). Но, что более интересно, мы обнаружили, что взаимодействия атомов углерода, расположенные в 5–11 координационных оболочках, вообще не важны. Действительно, кривые активности, рассчитанные с учетом C–C взаимодействий как в 11, так и только в четырех координатных оболочках, почти совпадают. Более того, можно отметить, что две различные параметризации [4, 5] энергий взаимодействия С–С дали нам одни и те же кривые активности, когда атомы углерода были расположены в первых четырех координатных оболочках. Поэтому анализ кривых активно- сти не позволяет получить однозначную информацию о взаимодействии С–С в ОЦК-железе.
Заключение
Мы провели обзор литературы по взаимодействию С–С и методам расчета углеродной активности в ОЦК-железе. Мы сравнили рассматриваемые данные с использованием Монте-Карло-моделирования углеродной активности в ОЦК-железе. Для ОЦК-железа было установлено, что для вычисления кривой активности углерода достаточно установить энергии взаимодействия для атомов углерода в первых четырех координационных сферах. Мы показали, что экспериментальная кривая активности может быть воспроизведена с использованием нескольких наборов параметров С–С взаимодействия. Поэтому проблема описания взаимодействия между атомами углерода математически некорректна.
Исследование выполнено за счет гранта Российского научного фонда №16-19-10252.
Список литературы Монте-Карло моделирование активности углерода в ОЦК-железе
- Могутнов, Б.М. Термодинамика железо-углеродистых сплавов / Б.М. Могутнов, Н.А. Томилин, Л.А. Шварцман. - М.: Металлургия, 1972. - 328 с.
- First-Principles Calculation of Effects of Carbon on Tetragonality and Magnetic Moment in Fe-C System / H. Ohtsuka, V.A. Dinh, T. Ohno et. al. // ISIJ International. - 2015. - Vol. 55, no. 11. - P. 2483-2491.
- Domain, C. Ab initio study of foreign interstitial atom (C, N) interactions with intrinsic point defects in α-Fe / C. Domain, C.S. Becquart, J. Foct // Physical Review B. - 2004. - Vol. 69. - P. 144112.
- Interaction between vacancies and interstitial solutes (C, N, and O) in α-Fe: From electronic structure to thermodynamic / C. Barouh, T. Schuler, C. Fu et. al. // Physical Review B. - 2014. - Vol. 90. - P. 054112.
- Ruban, A. Self-trapping of carbon atoms in α-Fe during the martensitic transformation: A qualitative picture from ab initio calculations / A. Ruban // Physical Review B. - 2014. - Vol. 90. - P. 144106.
- Ридный, Я.М. Первопринципное компьютерное моделирование взаимодействия атомов углерода на различных расстояниях в ОЦК- и ОЦТ-решетках феррита и мартенсита / Я.М. Ридный, А.А. Мирзоев, В.М. Счастливцев и др. // Физика металлов и металловедение. - 2018. - Т. 119, №. 6. - С. 611-617.
- Shiflet, G.J. A re-examination of the thermodynamics of the proeutectoid ferrite transformation in Fe-C alloys / G.J. Shiflet, J.R. Bradley, H.I. Aaronson // Metallurgical Transactions A. - 1978. - Vol. 9, no. 7. - P. 999-1008.
- Yiwen, M. C-C interaction energy in Fe-C alloys / M. Yiwen, T.Y. Hsu // Acta Metallurgica. - 1986. - Vol. 34, no. 2. - P. 325-331.
- McLellan, R.B. The C-C interaction energy in iron-carbon solid solutions / R.B. McLellan, C. Ko // Acta Metallurgica. - 1987. - Vol. 35, no. 8. - P. 2151-2156.
- Bhadeshia, H.K.D.H. Carbon-carbon interactions in iron / H.K.D.H. Bhadeshia // Journal of materials science. - 2004. - Vol. 39, no. 12. - P. 3949-3955.
- Murch, G.E. Computer simulation of the carbon activity in austenite / G.E. Murch, R.J. Thorn // Acta Metallurgica. - 1979. - Vol. 27, iss. 2. - P. 201-204.
- Sozinov, A.L. C-C interaction in iron-base austenite and interpretation of mossbauer spectra / A.L. Sozinov, A.G. Balanyuk, V.G. Gavriljuk // Acta Materialia. - 1997. - Vol. 45, no. 1. - P. 225-232.
- Gustafson, P. A thermodynamic evaluation of the C-Fe-W system / P. Gustafson // Metallurgical and Materials Transactions A. - 1987. - Vol. 18, iss. 2. - P. 259-267.
- Взаимодействие между атомами углерода и активность углерода в ГЦК-железе: термодинамические теории и компьютерное моделирование / Я.М. Ридный, А.А. Мирзоев, В.М. Счастливцев и др. // Физика металлов и металловедение. - 2018. - Т. 119, № 3. - С. 264-270.
- Ридный, Я.М. Ab initio моделирование энергии растворения и активности углерода в ГЦК-Fe / Я.М. Ридный, А.А. Мирзоев, Д.А. Мирзаев // Физика твёрдого тела. - 2017. - Т. 59, № 7. - C. 1255-1260.
- Lobo, J. Thermodynamics and Solubility of Carbon in Ferrite and Ferritic Fe-Mo Alloys / J. Lobo, G. Geiger // Metallurgical Transactions A. - 1976. - Vol. 7A. - P. 1347-1357.
