Морфология и маркеры иммобилизирующего интерстициального фиброза сердца

Автор: Шевченко Ю.Л., Плотницкий А.В., Судиловская В.В., Дубова Е.Д., Ульбашев Д.С.
Журнал: Вестник Национального медико-хирургического центра им. Н.И. Пирогова @vestnik-pirogov-center
Рубрика: Оригинальные статьи
Статья в выпуске: 3 т.17, 2022 года.
Бесплатный доступ
Фиброз-важнейший компонент восстановления целостности тканей и органов после их повреждения. В сердце же это фундаментальный этап в процессе его ремоделирования и, одновременно, центральное звено развития и прогрессирования хронической недостаточности кровообращения. Известно, что основной причиной сердечной недостаточности является дисфункция кардиомиоцитов, прежде всего, в результате ИБС. Однако нередко встречаются пациенты без явных признаков поражения собственно миокарда. Наша многолетняя клиническая практика, многочисленные научно-экспериментальные исследования позволили предположить, что причиной этого феномена является механический фактор, находящийся за пределами миокардиальных пучков здоровых кардиомиоцитов, но структурно тесно связанный с ними, - межуточная соединительная ткань с измененными физическими свойствами. Этот феномен назван иммобилизирующим интерстициальным фиброзом сердца. Цель: изучить морфологию соединительной ткани и экспрессию различных фиброгенных маркеров при иммобилизирующем интерстициальном фиброзе сердца. Материалы и методы. Исследование выполнено на базе патологоанатомического отделения НМХЦ им Н.И. Пирогова. I группа (n = 30)-пациенты с иммобилизирующим интерстициальным фиброзом сердца, причиной смерти которых была сердечная недостаточность. II (n = 10)-группа сравнения (без заболеваний сердечно-сосудистой системы). Возраст пациентов: 68±6,5 лет (I группа), 29±5,1 лет (II группа). Масса сердца: 486±141 г (I группа), 322±32 г (II группа). Гистологические срезы окрашивались гематоксилином и эозином, по Ван Гизону, Вейгерту и по Массону (метод трихром). Проводился иммуногистохимический анализ. Результаты. Между группами выявлены статистические различия по площади общей зоны фиброза: 13,7±7,4% (I группа), 5,6±4,2% (II группа), p = 0,001; экспрессии ММП-9: 14691±5256 в 1 мм2 (I группа), 7116±2831 в 1 мм2 (II группа), p=0,0001. Тенасцин-C определялся у пациентов с начальной стадией интерстициального фиброза. В группе сравнения экспрессия тенасцина-С и Bcl-2 не выявлялась. Исследуемые группы отличались по количеству обнаруженного коннексина-43: 24724±14764 в 1 мм2 (I группа), 38228±13548 в 1 мм2 (II группа), p = 0,02; фибронектина: 3354±719 в 1 мм2 (I группа), 1635±557 в 1 мм2 (II группа), (р = 0,00003). Выявлены значимые различия между группами в объеме волокон коллагена I типа: 4673±1292 в 1 мм2 (I группа), 2269±887 в 1 мм2 (II группа), р = 0,0001; III типа: 6959±1385 в 1 мм2 (I группа), 2566±568 в 1 мм2 (II группа), р = 0,00001. Заключение. Расшифровка молекулярных и структурных основ перестройки миокарда при фиброзе является ключом к пониманию патогенетических основ развития сердечной недостаточности. В основе иммобилизирующего интерстициального фиброза сердца лежит процесс изменения соединительнотканного каркаса, ведущий к увеличению его плотности, ограничению функции кардиомиоцитов. Иммуногистохимическое исследование и определение маркеров фиброзной перестройки сердца может позволить улучшить диагностику на ранних этапах развития заболевания и предотвратить его прогрессирование.
Иммобилизация, интерстициальный фиброз сердца, маркеры, сердечная недостаточность
Короткий адрес: https://sciup.org/140296591
IDR: 140296591 | DOI: 10.25881/20728255_2022_17_3_84
The morphology and markers of the immobilizing interstitial fibrosis of the heart
The fibrosis is the most important component of restoring the integrity of tissues and organs after their damage. In the heart, this is a fundamental stage in the process of its remodeling and, at the same time, the central link in the development and progression of chronic circulatory insufficiency. It is known that the main cause of heart failure is cardiomyocyte dysfunction, primarily as a result of coronary heart disease. However, patients often occur without obvious signs of myocardial damage proper. Our long-term clinical practice, numerous scientific and experimental studies allowed us to suggest that the cause of this phenomenon is a mechanical factor located outside the myocardial bundles of healthy cardiomyocytes, but structurally closely related to them-the interstitial connective tissue with altered physical properties. This phenomenon is called the immobilizing interstitial fibrosis of the heart. Objective: to study the morphology of connective tissue and the expression of various fibrogenic markers in immobilizing interstitial fibrosis of the heart. Materials and methods. The study was performed on the basis of the pathoanatomical department of the N.I. Pirogov NMHC. Group I (n = 30) - patients with immobilizing interstitial fibrosis of the heart, whose cause of death was heart failure. II (n = 10) is a comparison group (without diseases of the cardiovascular system). Age of patients: 68±6.5 years (group I), 29±5.1 years (group II). Heart weight: 486±141 g (group I), 322±32 g (group II). Histological sections were stained with hematoxylin and eosin, according to Van Gieson, Weigert and Masson (trichrome method). An immunohistochemical analysis was performed. Results. Statistical differences in the area of the common fibrosis zone were revealed between the groups: 13.7±7.4% (group I), 5.6±4.2% (group II), p = 0.001; MMP-9 expression: 14691±5256 in 1 mm2 (group I), 7116±2831 in 1 mm2 (group II), p = 0.0001. Tenascin-C was determined in patients with the initial stage of the interstitial fibrosis. In the comparison group, the expression of tenascin-C and Bcl-2 was not detected. The studied groups differed in the amount of detected connexin-43: 24724±14764 in 1 mm2 (group I), 38228±13548 in 1 mm2 (group II), p = 0.02; fibronectin: 3354±719 in 1 mm2 (group i), 1635±557 in 1 mm2 (group II), (p = 0.00003). Significant differences between the groups in the volume of collagen fibers of type I were revealed: 4673±1292 in 1 mm2 (group I), 2269±887 in 1 mm2 (group II), p = 0.0001; type III: 6959±1385 in 1 mm2 (group I), 2566±568 in 1 mm2 (II group), p = 0.00001. Conclusion. Deciphering the molecular and structural foundations of myocardial rearrangement in fibrosis is the key to understanding the pathogenetic foundations of the development of heart failure. The immobilizing interstitial fibrosis of the heart is based on the process of changing the connective tissue framework, leading to an increase in its density, limiting the function of cardiomyocytes. Immunohistochemi-cal examination and determination of markers of fibrous rearrangement of the heart can improve the diagnosis at the early stages of the development of the disease and prevent its progression.
Текст научной статьи Морфология и маркеры иммобилизирующего интерстициального фиброза сердца
Сердечная недостаточность (СН) — главная причина смертности во всех странах мира [1; 2]. Известно, что в основе ее лежит дисфункция кардиомиоцитов в результате ишемической болезни сердца, различных воспалительных процессов, чрезмерных физических перегрузок, в том числе при врожденных и приобретенных пороках, нарушений метаболизма с электролитными расстройствами, интокси-

каций, ушибов сердца, опухолей и прочее. Однако нередко встречаются пациенты с выраженной сердечной недостаточностью при отсутствии вышеперечисленных причин и явных признаков поражения собственно миокарда. Наша многолетняя клиническая практика, многочисленные научно-экспериментальные исследования позволили предположить, что причиной этого феномена является механический фактор, находящийся за пределами миокардиальных пучков здоровых кардиомиоцитов, но структурно тесно связанный с ними, — межуточная соединительная ткань с измененными физическими свойствами. Этот феномен назван иммобилизирующим интерстициальным фиброзом сердца [3].
Фиброгенез обусловлен, прежде всего, миофибробластами, образующимися в результате активации фибробластов, а также в процессе эпителиально-мезенхимальной трансформации [4–8]. Кроме того, определенную роль в нем играют макрофаги, моноциты, Т-лимфоциты, тучные клетки и эндотелиоциты. Миофибробласты под воздействием фиброгенных медиаторов синтезируют волокна коллагена, фибронектин и профибротические медиаторы, изменяя структуру внеклеточного матрикса. Основными фиброгенными медиаторами являются матриксные металлопротеиназы (ММП) и их тканевые ингибиторы (ТИММП), а также трансформирующий фактор роста β (TGF-β) [9–11]. Увеличение количества коллагена I и III типов приводит к изменениям структурной организации эндомизия и перимизия, нарушению электромеханических свойств сердца, иммобилизации неповрежденных кардиомиоцитов, что снижает общую сократительную способность левого желудочка (ЛЖ) [12]. Таким образом, расшифровка молекулярных и структурных элементов перестройки миокарда при иммобилизирующем интерстициальном фиброзе сердца является ключом к пониманию патогенетических основ развития СН.
Материалы и методы
Исследование выполнено на основе данных аутоп-сийного материала 40 пациентов, которые разделены на две группы: I группа (n = 30) — пациенты с иммобилизирующим интерстициальным фиброзом сердца, причиной смерти которых была сердечная недостаточность. II (n = 10) — группа сравнения (без заболеваний сердечнососудистой системы). В I группе средний возраст пациентов составил 68±6,5 лет, во II группе — 29±5,1 лет. В I группе масса сердца — 486±141 г, во II группе — 322±32 г.
Гистологический метод
Для морфологического исследования материал фиксирован в 10% нейтральном забуференном формалине непосредственно после изъятия. Гистологическая проводка осуществлена в вакуумном аппарате стандартным методом с использованием изопропила и этанола. Материал залит в блоки гомогенизированного парафина толщиной 5 мм. Гистологические срезы толщиной 4–5 мкм изготовлены непосредственно перед окрашиванием гематоксилином и эозином и проведением иммуногистохимических реакций. Монтирование гистологических срезов проведено на положительно заряженные стекла с высушиванием в соответствии с рекомендациями производителя. Полученные срезы депарафинировались и окрашивались гематоксилином и эозином по стандартной методике. Окрашивание срезов по Ван Гизону и по Массону (метод трихром) проводилось с использованием готовых наборов (Bio-Optica, Италия) в соответствии с прилагаемой инструкцией.
Иммуногистохимический метод
Иммуногистохимическое окрашивание выполнено автоматизированным методом с помощью иммуногистостейнера Ventana BenchMark Ultra с депарафинизацией и демаскировкой в аппарате. Была изучена экспрессия MMП-2, MMП-9, тенасцина-C, Bcl-2, коннексина-43, коллагенов I и III типов, TGF-β. Во всех случаях использовалась стандартизированная система детекции с бензидиновой меткой и непрямым методом выявления ultraVIEWDAB без дополнительного усиления сигнала.
Морфометрический метод
Интенсивность экспрессии указанных маркеров оценивалась автоматически с использованием платформы QuPath v0.2.3 по 4-х балльной шкале, где 0 баллов — отсутствие проявления, а 3 балла — выраженная экспрессия. Число позитивных клеток было подсчитано в участках произвольного размера с последующим подсчетом среднего значения позитивных клеток в 1 мм2 площади. Иллюстративный материал получен с помощью гистологического сканера Ventana iScan HT (Roche Diagnostics).
Статистический метод. Для статистической обработки данных использовали программный пакет «Statistica 10». Нормальность распределения оценивалась с помощью критериев Шапиро-Уилка, Колмогорова-Смирнова. Учитывая отличное от нормального распределения всех количественных показателей, анализ выполнялся с помощью непараметрического U-критерия Манна-Уитни. Результаты исследования представлены как среднее арифметическое ± стандартное отклонение (M±σ). За критический уровень значимости (p) при проверке гипотез было принято значение 0,01.
Результаты исследования
Объем зоны фиброза
На препаратах, окрашенных по методам Вейгерта, Массона и Ван-Гизона, была оценена средняя площадь зон фиброза (% отношения участков фиброза к общей площади исследованного фрагмента ткани): в I группе составила 13,7±7,4%, во II группе — 5,6±4,2% (p = 0,001) (Рис. 1–3). Оказалось, что с диффузным увеличением объема интерстициального фиброза в первой группе, происходило сдавление функционально активных кардиомиоцитов — их иммобилизация.
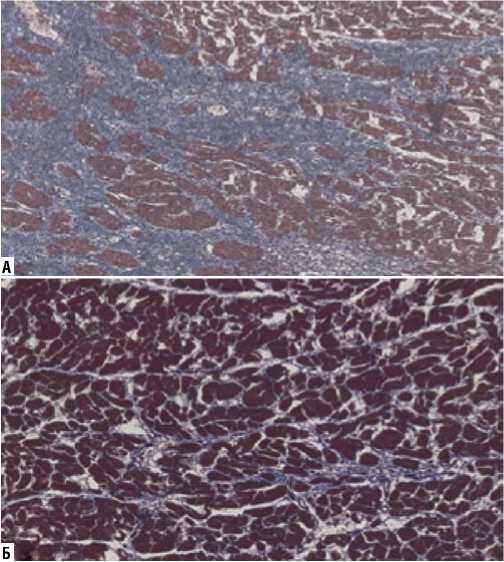
Рис. 2. Гистологические препараты миокарда (окраска по Массону (трихром), увеличение х200). А — I группа (участки иммобилизации кардиомиоцитов соединительнотканным каркасом); Б — II группа.

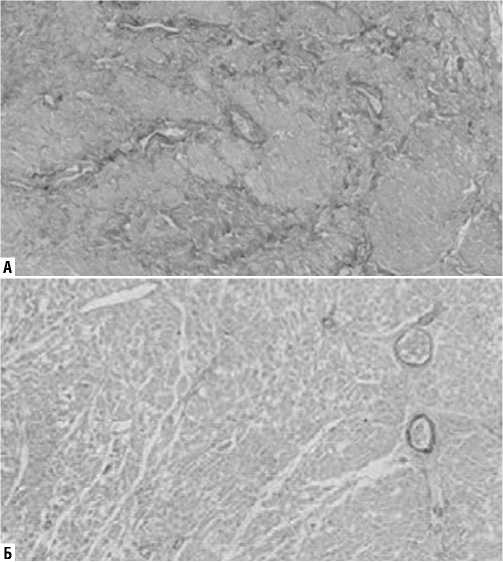
Рис. 1. Гистологические препараты миокарда (окраска по Вейгерту, увеличение х100). А — I группа (значимое увеличение объема соединительной ткани); Б — II группа.
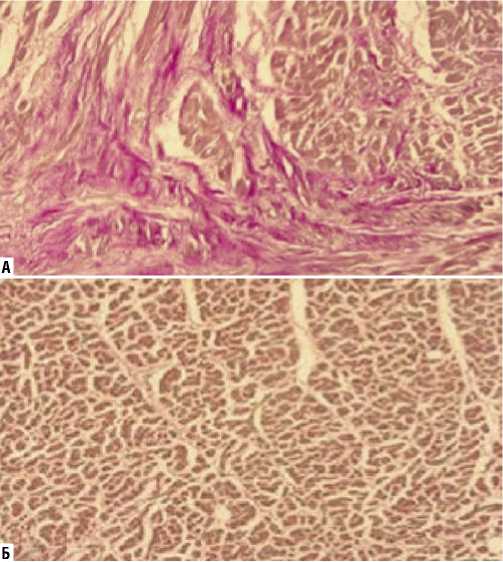
Рис. 3. Гистологические препараты миокарда (окраска по Ван Гизону, увеличение х200). А — I группа (выраженный интерстициальный фиброз); Б — II группа.
В первой группе в крайне тяжелой стадии иммобилизирующего интерстициального фиброза сердца
Рис. 4. Стадия коронарной ангиопатии — крайне тяжелая степень фиброза с констрикцией периферического коронарного русла (окраска гематоксилином и эозином, увеличение х200).
выявлено экстравазальное сдавление артериол миокарда с развитием значимой коронарной недостаточности, как итог, инфаркта миокарда (Рис. 4).
Металлопротеиназы
При иммуногистохимическом исследовании установлено, что экспрессия матриксных металлопротеиназ (MMП-2 и MMП-9) в миокарде определяется в цитоплазме кардиомиоцитов, эндотелиоцитов и фибробластов интерстициальной ткани. Для оценки уровня их экспрессии были выбраны кардиомиоциты и фибробласты
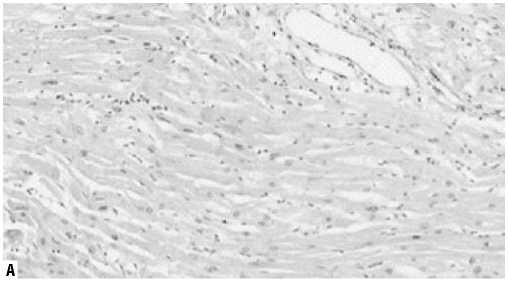
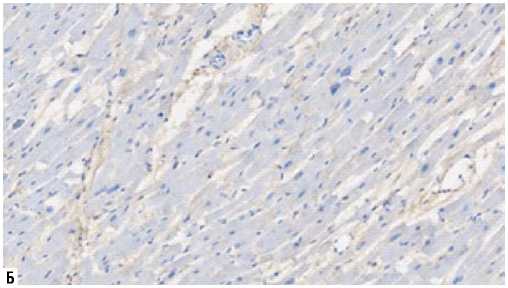
Рис. 5. Экспрессия ММП-2 (иммуногистохимическое исследование, увеличение х200). А — I группа, Б — II группа.
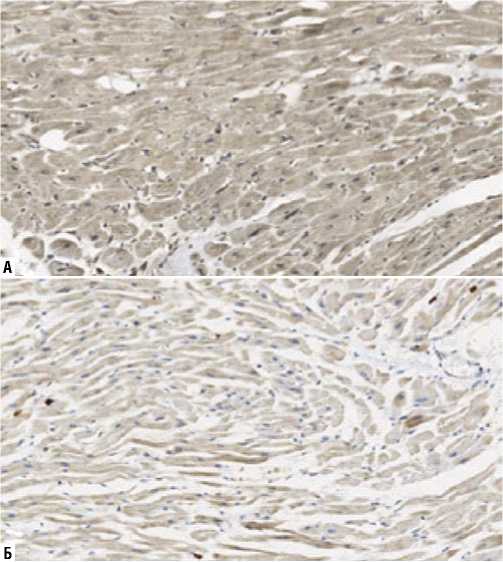
Рис. 6. Экспрессия ММП-9 (иммуногистохимическое исследование, увеличение х200). А — I группа; Б — II группа.
как основные клетки-мишени при ремоделировании миокарда. Среднее количество MMП-2 составило в I группе: 12710±5190 в 1 мм2, во II группе — 9804±7233 в 1 мм2 (p = 0,06) (Рис. 5).
Выявлено значимое различие между группами в экспрессии металлопротеиназы-9. Среднее количество MMП-9 кардиомиоцитов и фибробластов в I группе — 14691±5256 в 1 мм2, во II группе — 7116±2831 в 1 мм2 (p = 0,0001) (Рис 6, 7).
Металлопротеиназы, особенно MMП-9, являются очень «чувствительными» маркерами начальных стадий фиброза. Их содержание начинает повышаться в первую очередь за счет появления в цитоплазме местных фи-брогистиоцитов и мигрирующих из сосудов клеточных элементов крови. Это свидетельствует о запущенном механизме необратимых изменений в данном участке миокарда и анонсирует его будущую глубокую структурную перестройку при иммобилизирующем интерстициальном фиброза сердца .
Тенасцин-C
В норме тенасцин-C не обнаруживался в здоровых тканях сердца взрослых, за исключением сухожильных хорд сосочковых мышц и у основания створок сердечных клапанов. Он индуцируется или подавляется в развитых тканях во время повреждения, ремоделирования и при новообразованиях. Непосредственно в сердце тенас-цин-C взаимодействует с фибронектином, происходит регулирование экспрессии матричных металлопроте-
28 000
26 000.
24 000.
22 000.
20 000.
18 000.
16 000.
14 000.
12 000.
10 000.
8 000 _
6 000 _
4 000 _

Группа 1

п Медиана
25-75% —г- Мин.-Макс.
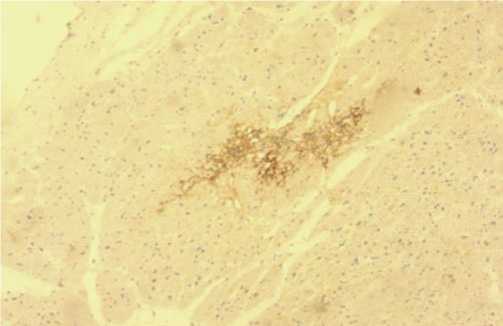
Рис. 8. Иммобилизирующий интерстициальный фиброз сердца, экспрессия тенасцина-С (иммуногистохимическое исследование, увеличение х100).
60 000
50 000.
Рис. 9. Иммобилизирующий интерстициальный фиброз сердца, экспрессия BCL-2 (иммуногистохимическое исследование, увеличение х100).
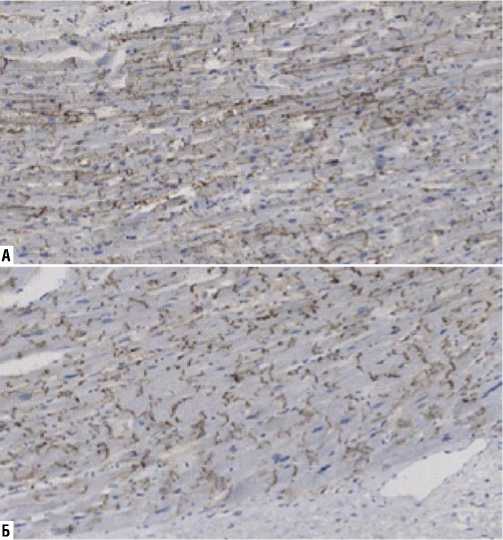
Рис. 10. Экспрессия коннексина-43 (иммуногистохимическое исследование, увеличение х200). А — I группа; Б — II группа.
40 000.
30 000.
Bcl-2
Bcl-2 (B-cell lymphoma 2) — белок, относящийся к семейству антиапоптотических протеинов. Он является ключевым фактором устойчивости сердечных фибробластов к запрограммированной гибели клеток. В свою очередь прямой механизм влияния на апоптоз осуществляется семейством белков BH3-only, которые инициализируются посредством высвобождения еще одного проапоптотического белка — цитохрома С. Устойчивость сердечных фибробластов/миофибро-бластов к митохондриально-зависимому апоптозу подчеркивает их повышенный потенциал в условиях гипоксии.
Этот белок обнаруживался в цитоплазме как кардиомиоцитов, так и фибробластических клеточных элементов в миокарде, прилежащих к зонам фиброза или вовлеченных в процесс. Экспрессия Bcl-2 у пациентов без установленного диагноза инфаркта миокарда может служить одним из критериев, указывающим на имеющуюся дисфункцию сердечной мышцы при нарушении ее трофики в результате прямого повреждения сосудов
20 000.
10 000.


О
Группа 1
Группа 2
° Медиана 25-75% Мин.-Макс.
Рис. 11. Ящичные диаграммы, отражающие статистическое различие в количестве определенного коннексина-43 в I и II группах.
или при механическом сдавлении этих зон измененной соединительной тканью (Рис. 9). Во II группе экспрессия Bcl-2, как и тенасцина-С не выявлялась.
Коннексин-43
Экспрессия коннексина-43 в I группе (24724±14764 в 1 мм2) определялась в меньшем количестве, в отличие от II группы (38228±13548 в 1 мм2) (p = 0,02). При иммобилизирующем интерстициальном фиброзе сердца происходила «латерализация» коннексина-43, тогда как
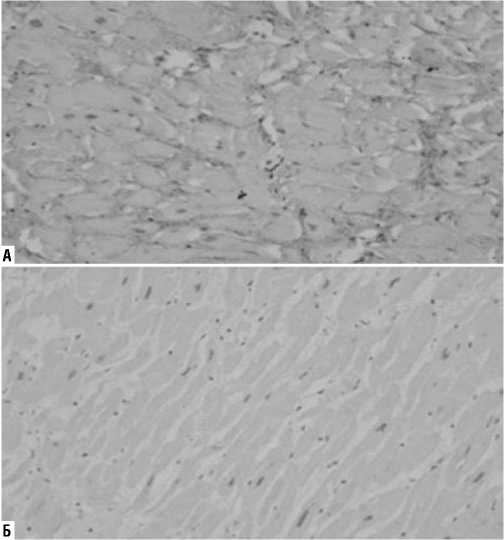
Рис. 12. Коллагеновые волокна I типа (окраска по Вейгерту, увеличение х400). А — I группа, хаотичное пересечение пучков, образующих замкнутые пространства вокруг отдельных кардиомиоцитов; Б — II группа, упорядоченное расположение волокон коллагена.
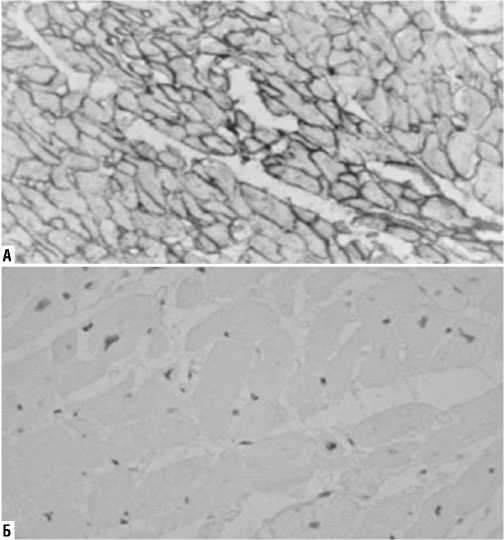
Рис. 14. Коллагеновые волокна III типа (окраска по Вейгерту, увеличение х400). А — I группа, неупорядоченное расположение, без четко различимых пространственных структур; Б — II группа, редкие пучки коллагеновых волокон.
8 000
7 000 _
6 000.
5 000.
4 000,
3 000 _I
2 000 _ —°
1 000-^—
Группа 1 Группа 2
□ Медиана 25-75% Мин.-Макс.
11 000
10 000 .
9 000 .
8 000 .
7 000 .
6 000 .
5 000 .
4 000 .
3 000 .
2 000 .
1 000..


° Медиана
25-75% Мин.-Макс.
Рис. 13. Ящичные диаграммы, отражающие статистическое различие в количестве определенного коллагена I типа в I и II группах.
Рис. 15. Ящичные диаграммы, отражающие статистическое различие в количестве определенного коллагена III типа в I и II группах.
у пациентов со здоровым миокардом коннексин-43 выявлялся преимущественно в зоне вставочных дисков кардиомиоцитов (Рис. 10, 11).
Коллаген I и III типов
Отмечалось значимое увеличение числа волокон коллагена I типа у пациентов с иммобилизирующим интерстициальным фиброзом сердца (4673±1292 в 1 мм2) по сравнению с группой сравнения (2269±887 в 1 мм2)
(p = 0,0001). Волокна располагались хаотично, формируя сетевидные структуры (Рис. 12, 13).
Количество коллагена III типа также было больше в I группе: 6959±1385 в 1 мм2 (I группа), 2566±568 в 1 мм2 (II группа) (p = 0,00001) (Рис. 14, 15).
Фибронектин
В группе аутопсий пациентов с иммобилизирующим интерстициальным фиброзом сердца отмечено
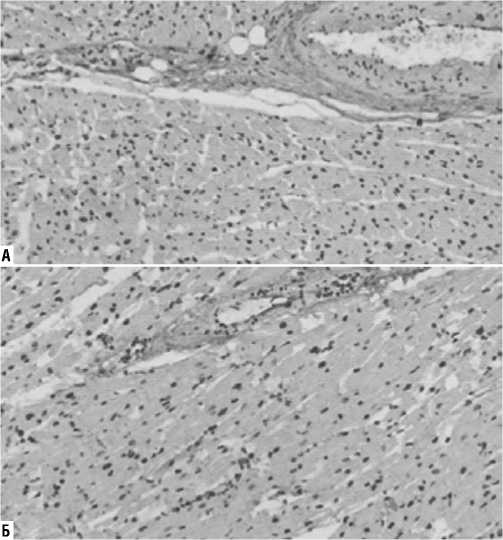
Рис. 16. Экспрессия фибронектина. А — I группа, Б — II группа (иммуногистохимическое исследование, увеличение х200).
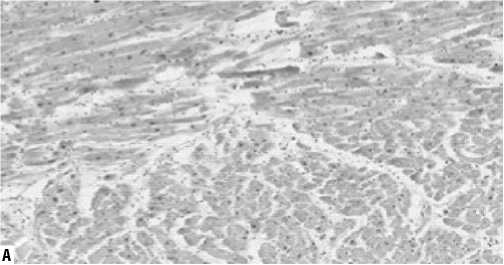
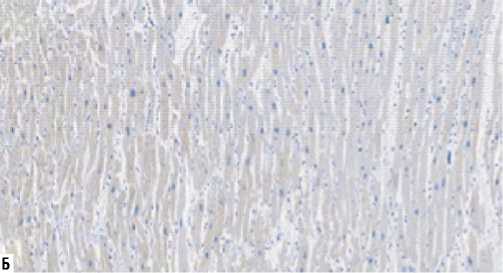
Рис. 18. Экспрессия TGF- β . А — I группа, Б — II группа (иммуногистохимическое исследование, увеличение х200).
5 000
4 500 .
4 000 .
3 500 .
3 000 .
2 500 .
2 000 .
1 500 .
1 000 _

7 000
6 000 .
5 000 .
4 000 .
3 000 .
2 000 .
1 000.
О
Группа 1
Группа 2
° Медиана
25-75% ~~i Мин.-Макс.
□ Медиана

Группа 1
25-75%

Группа 2
Рис. 17. Ящичные диаграммы, отражающие статистическое различие в количестве определенного фибронектина в I и II группах.
Рис. 19. Ящичные диаграммы, отражающие статистическое различие в количестве определенного TGF- β в I и II группах.
значимое повышение экспрессии фибронектина миофибробластами и фибробластами (3354±719 в 1 мм2) по сравнению со второй группой, где она была слабо выражена (1635±557 в 1 мм2), (p = 0,00003) (Рис. 16, 17).
TGF-β
Выявлены различия в экспрессии TGF-β: в I группе — 4837±1161 в 1 мм2 и во II — 2646±1682 в 1 мм2 (p = 0,001) (Рис. 18, 19).
Обсуждение
В ответ на повреждение и воздействие различных факторов фибробласты интерстициальной ткани сердца и иммунные клетки начинают перестраивать внеклеточный матрикс путем выделения ММП и ТИМПП [12; 13]. Они напрямую влияют на обновление и гомеостаз внеклеточного матрикса. MMП-9 выделяется преимущественно лейкоцитами интерстиция, при этом уровень его экспрессии кардиомиоцитами минимален [14], повышается с возрастом, причем это касается как уровня экспрессии

м-РНК MMП-9, так и самого белка в плазме крови и тканях миокарда ЛЖ [15; 16]. Изменение количества ММП и ТИМПП, по данным различных исследований, отмечено при, так называемой, идиопатической дилатационной кардиомиопатии [17]. А также у пациентов с СН на фоне артериальной гипертензии [18]. Подавление MMП-9 зависимого пути уменьшает процесс фиброгенеза в сердце [19].
Выявленное повышение экспрессии MMП-9 свидетельствует о глубокой перестройке межуточной соединительной ткани со значительными нарушениями ее структуры и функции при иммобилизирующем интерстициальном фиброзе, когда отмечаются выраженные диффузные склеротические изменения, увеличение в объеме эндомизия, перимизия и сдавление кардиомиоцитов.
Экспрессия тенасцина-C в миокарде может свидетельствовать о начальных стадиях процесса так же, как и увеличение антиапоптотических белков Bcl-2.
Нексусы в сердце взрослого человека входят в структуру вставочных дисков, находятся между десмосомами и плотными контактами (кроме того, небольшая часть контактов этого типа расположена на боковой мембране кардиомиоцита). У новорожденного щелевые контакты распределены в структуре вставочных дисков хаотично, однако по мере созревания сердечной ткани они перемещаются в определённые зоны дисков, формируя скопления с низким уровнем сопротивления электрическому импульсу [20]. Анизотропное распределение скоплений щелевых контактов обеспечивает преимущественно продольную передачу импульса и синхронизированное сокращение миокарда [21]. Структурной основой нексусов являются 2 типа белков: коннексины и паннексины. Функция паннексинов изучена плохо, при этом в последнее время большое внимание уделяется изучению роли коннексинов в развитии сердечной патологии. Каждый отдел сердца содержит различные типы коннексинов с разными функциями, которые влияют на проводимость контакта и его проницаемость для ионов Na+, определяя объем импульса [21]. Идентифицирован 21 ген, кодирующий синтез различных типов коннексинов. Каждая молекула коннексина представляет собой комплекс из 4 трансмембранных доменов, 2 внеклеточных и 1 внутриклеточной петли, а также расположенных в цитоплазме амино- и карбоксильных терминальных групп [22]. Самый распространенный тип в сердце, который экспрессируется во всех его отделах, включая проводящую систему, — коннексин-43. В норме он расположен во вставочных дисках, небольшое его количество также определяется в парануклеарной зоне цитоплазмы кардиомиоцитов (эндоплазматическая сеть) [23].
Увеличение экспрессии коннексина-43 отмечается при компенсированной гипертрофии сердца, при декомпенсации ее уровень падает [24; 25]. Снижение также происходит с возрастом [26; 27]. В зоне выраженного фиброза его определяется меньше, происходит «латера-лизация» коннексина-43 — это частично связано с нару- шением синтеза NO и повышение активности MMП [25; 28]. Многочисленные экспериментальные и клинические исследования продемонстрировали изменения экспрессии коннексина-43 при разных сердечно-сосудистых патологиях [29–34].
Полученные результаты свидетельствуют о снижении экспрессии коннексина-43 кардиомиоцитами и его перераспределении в клетке у пациентов с тяжелой стадией иммобилизирующего интерстициального фиброза миокарда за счет нарушения щелевых контактов.
Основной морфологический субстрат фиброза миокарда — увеличение количества коллагенов I и III типов [35]. 80–90% всех коллагенов в организме представлено I типом [36; 37]. Коллаген III типа составляет до 10% [38], при этом распределение и соотношение коллагенов I и III типов определяет основные физические свойства фиброзированной ткани (ее плотность) [9; 39].
Количество коллагенов I и III типов в интерстиции миокарда значительно повышается с возрастом (как в экспериментальных моделях, так и в клинике), по некоторым данным такое повышение происходит практически в два раза [40]. Кроме того, с возрастом изменяется соотношение основных типов коллагенов (количество волокон III типа снижается, тогда как I — повышается). Именно это соотношение является ключевым в оценке «жесткости» миокарда [40,41]. Радикально изменяется кардиодинамика (вначале появляется дисфункция диастолическая, а потом и систолическая), нарушается эффект спиральной «закрутки» сердца, в итоге усугубляется СН [42].
В исследовании у пациентов с иммобилизирующим интерстициальным фиброзом сердца значимо увеличивалось содержание коллагена I типа, менялась его пространственная структура — образовывались сетки, пересекающиеся пучки, формировались замкнутые пространства вокруг отдельных кардиомиоцитов. Такой каркас также включал более многочисленные пучки волокон коллагена III типа, расположенные хаотично без четко различимых пространственных структур. Все это приводило к ухудшению сократительной функции миокарда.
Фибронектин — самоорганизующийся комплексный гликопротеин, вырабатываемый фибробластами и миофибробластами в ответ на профибротические стимулы. Фибронектин связывается с коллагенами, интегринами, протегликанами и другими молекулами межклеточного пространства, регулируя их структурные перестройки [43; 44]. Его экспрессия повышается при различных типах кардиосклероза, в том числе и при семейной форме неишемической дилатационной кардиомиопатии [45–50]. Данные исследования демонстрируют увеличение его экспрессии у пациентов с иммобилизирующим интерстициальным фиброзом сердца .
Один из ключевых фиброгенных цитокинов — TGF-β выделяется активированными миофибробластами, а также клетками, участвующими в воспалительных процессах. TGF-β — это белок-цитокин,
контролирующий процессы дифференцировки и пролиферации большинства клеток организма. В зонах повреждения миокарда TGF-β стимулирует синтез фибронектина посредством активации сигнального пути Smad, а также последующее накопление коллагенов I и III типа, обусловленного TGF-β-Smad-активированными протеинкиназами [38]. Кроме того, TGF-β подавляет воспалительный ответ в зоне повреждения и способствует гипертрофии кардиомиоцитов [38; 46; 51; 52]. Полученные данные свидетельствуют о значимом повышении экспрессии TGF-β в группе пациентов с иммобилизирующим интерстициальным фиброзом сердца , этот цитокин стимулировал синтез соединительной ткани, что приводило к увеличению ригидности и снижению сократимости миокарда.
Заключение
Таким образом, расшифровка молекулярных и структурных основ перестройки миокарда при фиброзе является ключом к пониманию патогенетических основ развития сердечной недостаточности.
В основе иммобилизирующего интерстициального фиброза сердца лежит процесс изменения соединительнотканного каркаса миокарда, увеличение его «жесткости» со сдавлением кардиомиоцитов и ограничением их сократительной функции.
Использование иммуногистохимического исследования и определение маркеров фиброзной перестройки может способствовать диагностике иммобилизирующего интерстициального фиброза сердца на его ранних этапах и возможности предотвращения усугубляющейся тяжести СН.
Авторы заявляют об отсутствии конфликта интересов (The authors declare no conflict of interest).
Список литературы Морфология и маркеры иммобилизирующего интерстициального фиброза сердца
- Bui AL, Horwich TB, Fonarow GC. Epidemiology and risk profile of heart failure. Nat Rev Cardiol. 2011; 8(1): 30-41. doi: 10.1038/nrcardio.2010.165.
- Giamouzis G, Triposkiadis F, Butler J, et al. Heart failure. Cardiol Res Pract. 2011; 2011: 159608. doi:10.4061/2011/159608.
- Шевченко Ю.Л. Иммобилизирующий интерстициальный фиброз сердца. Вестник НМХЦ им. Н.И. Пирогова. — 2022. — Т.17. — №2. — С. 4-10. [Shevchenko YuL. The immobilizing interstitial fibrosis of the heart. Vestnik NMHC im. N.I. Pirogova. 2022; 17(2): 4-10. (In Russ).] doi:10.25881 /20728255_2022_17_2_4.
- Swierblewska E, Wolf J, Kunicka K, et al. Prevalence and distribution of left ventricular diastolic dysfunction in treated patients with long-lasting hypertension. Blood Press. 2018; 27(6): 376-384. doi: 10.1080/08037051.2018.1484661.
- Jens van de Wouw J, Broekhuizen M, Sorop O, et al. Chronic Kidney Disease as a Risk Factor for Heart Failure With Preserved Ejection Fraction: A Focus on Microcirculatory Factors and Therapeutic Targets. Front Physiol. 2019; 10: 1108. doi: 10.3389/fphys.2019.01108.
- Riet EE, Hoes AW, Wagenaar KP, et al. Epidemiology of heart failure: the prevalence of heart failure and ventricular dysfunction in older adults over time. A systematic review. Eur J Heart Fail. 2016; 18(3): 242-52. doi: 10.1002/ejhf.483.
- Mocan M, Mocan Hognogi LD, Anton FP, et al. Biomarkers of Inflammation in Left Ventricular Diastolic Dysfunction. Dis Markers. 2019; 2019: 7583690. doi: 10.1155/2019/7583690.
- De Boer RA, De Keulenaer G, Bauersachs J, et al. Towards better definition, quantification and treatment of fibrosis in heart failure. A scientific roadmap by the Committee of Translational Research of the Heart Failure Association (HFA) of the European Society of Cardiology. Eur J Heart Fail. 2019; 21(3): 272-285. doi: 10.1002/ejhf.1406.
- Suthahar N, Meijers WC, Sillje HHW, de Boer RA. From Inflammation to Fibrosis-Molecular and Cellular Mechanisms of Myocardial Tissue Remodelling and Perspectives on Differential Treatment Opportunities. Curr Heart Fail Rep. 2017; 14(4): 235-250. doi: 10.1007/s11897-017-0343-y.
- Legere SA, Haidl ID, Legare JF, Marshall JS. Mast Cells in Cardiac Fibrosis: New Insights Suggest Opportunities for Intervention. Front Immunol. 2019; 10: 580. doi: 10.3389/fimmu.2019.00580.
- Nevers T, Salvador AM, Velazquez F, et al. Th1 effector T cells selectively orchestrate cardiac fibrosis in nonischemic heart failure. J Exp Med. 2017; 214(11): 3311-3329. doi: 10.1084/jem.20161791.
- Piek A, de Boer RA, Sillje HH. The fibrosis-cell death axis in heart failure. Heart Fail Rev. 2016; 21(2): 199-211. doi: 10.1007/s10741-016-9536-9.
- Frangogiannis NG. The extracellular matrix in myocardial injury, repair, and remodeling. J Clin Invest. 2017;127(5):1600-1612. doi: 10.1172/ JCI87491.
- Huet E, Gabison E, Vallee B, et al. Deletion of extracellular matrix metal-loproteinase inducer/CD147 induces altered cardiac extracellular matrix remodeling in aging mice. J Physiol Pharmacol. 2015; 66(3): 355-66.
- Chiao YA, Dai Q, Zhang J, et al. Multi-analyte profiling reveals matrix meta-lloproteinase-9 and monocyte chemotactic protein-1 as plasma biomarkers of cardiac aging. Circ Cardiovasc Genet. 2011; 4(4): 455-62. doi: 10.1161/ CIRCGENETICS.111.959981.
- Meschiari CA, Ero OK, Pan H, et al. The impact of aging on cardiac extracellular matrix. Geroscience. 2017; 39(1): 7-18. doi: 10.1007/s11357-017-9959-9.
- Antonov IB, Kozlov KL, Pal'tseva EM, et al. Matrix Metalloproteinases MMP-1 and MMP-9 and Their Inhibitor TIMP-1 as Markers of Dilated Cardiomyopathy in Patients of Different Age. Bull Exp Biol Med. 2018; 164(4): 550-553. doi: 10.1007/s10517-018-4030-0.
- Martos R, Baugh J, Ledwidge M, et al. Diagnosis of heart failure with preserved ejection fraction: improved accuracy with the use of markers of collagen turnover. Eur J Heart Fail. 2009; 11(2): 191-7. doi: 10.1093/eurjhf/ hfn036.
- Wang JH, Su F, Wang S, et al. CXCR6 deficiency attenuates pressure overload-induced monocytes migration and cardiac fibrosis through dow-nregulating TNF-a-dependent MMP-9 pathway. Int J Clin Exp Pathol. 2014; 7(10): 6514-23.
- Gutstein DE, Liu FY, Meyers MB, Choo A, Fishman GI. The organization of adherens junctions and desmosomes at the cardiac intercalated disc is independent of gap junctions. J Cell Sci. 2003; 116(Pt5): 875-85. doi: 10.1242/jcs.00258.
- Duffy HS, Fort AG, Spray DC. Cardiac connexins: genes to nexus. Adv Cardiol. 2006; 42: 1-17. doi: 10.1159/000092550.
- Solan JL, Lampe PD. Connexin phosphorylation as a regulatory event linked to gap junction channel assembly. Biochim Biophys Acta. 2005; 1711(2): 154-63. doi: 10.1016/j.bbamem.2004.09.013.
- Lampe PD, Cooper CD, King TJ, Burt JM. Analysis of Connexin43 phosp-horylated at S325, S328 and S330 in normoxic and ischemic heart. J. Cell Sci. 2006; 119: 3435-3442.
- Kostin S, Dammer S, Hein S, et al. Connexin 43 expression and distribution in compensated and decompensated cardiac hypertrophy in patients with aortic stenosis. Cardiovasc Res. 2004; 62(2): 426-36. doi: 10.1016/j.cardi-ores.2003.12.010.
- Boengler K, Schulz R. Connexin 43 and Mitochondria in Cardiovascular Health and Disease. Adv Exp Med Biol. 2017; 982: 227-246. doi: 10.1007/978-3-319-55330-6_12.
- Kostin S, Rieger M, Dammer S, et al. Gap junction remodeling and altered connexin43 expression in the failing human heart. Mol Cell Biochem. 2003; 242(1-2): 135-44.
- Fannin J, Rice KM, Thulluri S, et al. Age-associated alterations of cardiac structure and function in the female F344xBN rat heart. Age (Dordr). 2014; 36(4): 9684. doi: 10.1007/s11357-014-9684-6.
- Givvimani S, Pushpakumar S, Veeranki S, Tyagi SC. Dysregulation of Mfn2 and Drp-1 proteins in heart failure. Can J Physiol Pharmacol. 2014; 92(7): 583-91. doi: 10.1139/cjpp-2014-0060.
- Ai X, Zhao W, Pogwizd SM. Connexin43 knockdown or overexpression modulates cell coupling in control and failing rabbit left ventricular myocytes. Cardiovasc Res. 2010; 85(4): 751-62. doi: 10.1093/cvr/cvp353.
- dos Santos DO, Blefari V, Prado FP, et al. Reduced expression of adherens and gap junction proteins can have a fundamental role in the development of heart failure following cardiac hypertrophy in rats. Exp Mol Pathol. 2016; 100(1): 167-76. doi: 10.1016/j.yexmp.2015.12.009.
- Dupont E, Matsushita T, Kaba RA, et al. Altered connexin expression in human congestive heart failure. J Mol Cell Cardiol. 2001; 33(2): 359-71. doi: 10.1006/jmcc.2000.1308.
- Kostin S, Klein G, Szalay Z, et al. Structural correlate of atrial fibrillation in human patients. Cardiovasc Res. 2002; 54(2): 361-79. doi: 10.1016/ s0008-6363(02)00273-0.
- Lousinha A, Pereira G, Borrecho G, et al. Atrial fibrosis and decreased connexin 43 in rat hearts after exposure to high-intensity infrasound. Exp Mol Pathol. 2020; 114: 104409. doi: 10.1016/j.yexmp.2020.104409.
- Baum JR, Long B, Cabo C, Duffy HS. Myofibroblasts cause heterogeneous Cx43 reduction and are unlikely to be coupled to myocytes in the healing canine infarct. Am J Physiol Heart Circ Physiol. 2012; 302(3): H790-800. doi: 10.1152/ajpheart.00498.2011.
- Nagao K, Inada T, Tamura A, et al. Circulating markers of collagen types I, III, and IV in patients with dilated cardiomyopathy: relationships with myocardial collagen expression. ESC Heart Fail. 2018; 5(6): 1044-1051. doi: 10.1002/ehf2.12360.
- Weber KT, Janicki JS, Shroff SG, et al. Collagen remodeling of the pressure-overloaded, hypertrophied nonhuman primate myocardium. Circ Res. 1988; 62(4): 757-65. doi: 10.1161/01.res.62.4.757.
- Suthahar N, Meijers WC, Silljé HHW, de Boer RA. From Inflammation to Fibrosis-Molecular and Cellular Mechanisms of Myocardial Tissue Remodelling and Perspectives on Differential Treatment Opportunities. Curr Heart Fail Rep. 2017; 14(4): 235-250. doi: 10.1007/s11897-017-0343-y.
- Weber KT, Sun Y, Bhattacharya SK, et al. Myofibroblast-mediated mechanisms of pathological remodelling of the heart. Nat Rev Cardiol. 2013; 10(1): 15-26. doi: 10.1038/nrcardio.2012.158.
- Newby AC. Metalloproteinase expression in monocytes and macrophages and its relationship to atherosclerotic plaque instability. Arterioscler Thro-mb Vasc Biol. 2008; 28(12): 2108-14. doi: 10.1161/ATVBAHA.108.173898.
- Gazoti Debessa CR, Mesiano Maifrino LB, Rodrigues de Souza R. Age related changes of the collagen network of the human heart. Mech Ageing Dev. 2001; 122(10): 1049-58. doi: 10.1016/s0047-6374(01)00238-x.
- Mendes AB, Ferro M, Rodrigues B, et al. Quantification of left ventricular myocardial collagen system in children, young adults, and the elderly. Medicina (B Aires). 2012; 72(3): 216-20.
- Meschiari CA, Pinheiro LC, Guimaraes DA, et al. Sodium nitrite attenuates MMP-9 production by endothelial cells and may explain similar effects of atorvastatin. Naunyn Schmiedebergs Arch Pharmacol. 2016; 389(2): 223-31. doi: 10.1007/s00210-015-1192-4.
- Halper J, Kjaer M. Basic components of connective tissues and extracellular matrix: elastin, fibrillin, fibulins, fibrinogen, fibronectin, laminin, tenascins and thrombospondins. Adv Exp Med Biol. 2014; 802: 31-47. doi: 10.1007/978-94-007-7893-1_3.
- Nattel S. Molecular and Cellular Mechanisms of Atrial Fibrosis in Atrial Fibrillation. JACC Clin Electrophysiol. 2017; 3(5): 425-435. doi: 10.1016/ j.jacep.2017.03.002.
- Cojan-Minzat BO, Zlibut A, Agoston-Coldea L. Non-ischemic dilated cardiomyopathy and cardiac fibrosis. Heart Fail Rev. 2020. doi: 10.1007/ s10741-020-09940-0.
- Ma ZG, Yuan YP, Wu HM, et al. Cardiac fibrosis: new insights into the pathogenesis. Int J Biol Sci. 2018; 14(12): 1645-1657. doi: 10.7150/ijbs.28103.
- Pedram A, Razandi M, Narayanan R, Levin ER. Estrogen receptor beta signals to inhibition of cardiac fibrosis. Mol Cell Endocrinol. 2016; 434: 57-68. doi: 10.1016/j.mce.2016.06.018.
- Medzikovic L, Aryan L, Eghbali M.J Connecting sex differences, estrogen signaling, and microRNAs in cardiac fibrosis. Mol Med (Berl). 2019; 97 (10): 1385-1398. doi: 10.1007/s00109-019-01833-6.
- Chatzifrangkeskou M, Le Dour C, Wu W, et al. ERK1/2 directly acts on CTGF/CCN2 expression to mediate myocardial fibrosis in cardiomyopathy caused by mutations in the lamin A/C gene. Hum Mol Genet. 2016; 25(11): 2220-2233. doi: 10.1093/hmg/ddw090.
- Levick SP, Widiapradja A. Mast Cells: Key Contributors to Cardiac Fibrosis. Int J Mol Sci. 2018; 19(1): 231. doi: 10.3390/ijms19010231.
- Gong D, Shi W, Yi SJ, et al. TGFß signaling plays a critical role in promoting alternative macrophage activation. BMC Immunol. 2012; 13: 31. doi: 10.1186/1471-2172-13-31.
- Kong P, Christia P, Frangogiannis NG. The pathogenesis of cardiac fibrosis. Cell Mol Life Sci. 2014; 71(4): 549-74. doi: 10.1007/s00018-013-1349-6.
