Мутации гена ламина A/C (LMNA) у пациентов с дилатационной кардиомиопатией и их фенотипические проявления
 у пациентов с дилатационной кардиомиопатией и их фенотипические проявления")
Автор: Вайханская Т.Г., Сивицкая Л.Н., Даниленко Н.Г., Курушко Т.В., Давыденко О.Г.
Журнал: Евразийский кардиологический журнал @eurasian-cardiology-journal
Рубрика: Оригинальные статьи
Статья в выпуске: 1, 2016 года.
Бесплатный доступ
В статье приведены современные представления о структуре и функциях ядерных белков ламинов, патологические механизмы генных ламиновых мутаций и лечебно-диагностические проблемы ламин-ассоциированных форм дила-тационной кардиомиопатии (ДКМП). ДКМП, обусловленная мутациями ядерного гена ламина (LMNA), часто связана с нарушениями сердечного ритма, проводимости и различными скелетно-мышечными расстройствами. Это заболевание отличается неблагоприятным прогнозом, обусловленным высоким риском внезапной сердечной смерти из-за нарушений проводимости (жизнеугрожающие блокады) или желудочковой тахиаритмии (фатальные желудочковые тахикардии/ фибрилляции). В статье представлены 2 клинических случая ламиновых фенотипов ДКМП, ассоциированных с мутациями гена LMNA. В обучающем аспекте для практических врачей изложены дифференциальные клинические признаки потенциальных носителей LMNA мутаций, так как генетическая диагностика ламиновых ДКМП позволяет своевременно определить оптимальную тактику лечения и необходимость профилактической имплантации кардиовертера-дефибрил-лятора.
Белки ядерной ламины, мутации гена ламина а/с (lmna), дилатационная кардиомиопатия (дкмп), жизнеугрожающие аритмии, кардиовертер-дефибриллятор (квд)
Короткий адрес: https://sciup.org/14342811
IDR: 14342811
Lamin A/C gene (LMNA) mutations in patients with dilated cardiomyopathy and their phenotypic manifestation
This article presents the current view on the structure and functions of nuclear lamin proteins, pathological phenotypes of persons with LMNA mutations and clinical problems of lamin-related dilated cardiomyopathy diagnostics and management. Dilated cardiomyopathy (DCM) caused by mutations in the LMNA gene is often associated with conduction disorders, cardiac arrhythmias and extracardiac features with discrete muscle disruption. This disease is characterized by a poor prognosis and high risk of sudden cardiac death due to conduction disturbances (life-threatening blockade) or ventricular tachyarrhythmias (fatal ventricular tachycardias\fibrillation). We describe here 2 cases of lamin-type DCM associated with definite LMNA mutations. Educational focus for reader-cardiologist is а recognition of potential carriers followed by molecular genetic testing for diagnostics of LMNA mutation and tissues regarding optimal management of patients, especially timing for prophylactic cardioverter-defibrillator.
Текст обзорной статьи Мутации гена ламина A/C (LMNA) у пациентов с дилатационной кардиомиопатией и их фенотипические проявления
|
Сведения об авторах: |
|
|
Cивицкая Лариса Николаевна |
ГНУ «Институт генетики и цитологии», НАН Беларуси, старший научный сотрудник лаборатории нехромосомной наследственности, канд. биол. наук Адрес: 220072, Беларусь, г. Минск, ул. Академическая 27, Тел.+375172686420, E- mail: cytoplasmic@mail.ru |
|
Даниленко Нина Генусовна |
ГНУ «Институт генетики и цитологии», НАН Беларуси, ведущий научный сотрудник лаборатории нехромосомной наследственности, канд. биол. наук, Адрес: 220072, Беларусь, г. Минск, ул. Академическая 27, Тел.+375172686420, E- mail: cytoplasmic@mail.ru |
|
Курушко Татьяна Валентиновна |
РНПЦ «Кардиология», врач отделения функциональной диагностики, 220036, Беларусь, г. Минск, ул. Р. Люксембург, 110, Тел.+375293506880, E- mail: tatkuko@mail.ru |
|
Давыденко Олег Георгиевич |
ГНУ «Институт генетики и цитологии», НАН Беларуси, д.б.н., член-корр. НАН Беларуси, заведующий лабораторией нехромосомной наследственности, Адрес: 220072, Беларусь, г. Минск, ул. Академическая, 27, Тел.+375172686420, E- mail: cytoplasmic@mail.ru |
|
Автор для контакта с редакцией: Вайханская Татьяна Геннадьевна |
РНПЦ «Кардиология», ведущий научный сотрудник функциональной группы клинической патофизиологии кровообращения, канд. мед. Наук, Адрес: 220036, Беларусь, г. Минск, ул. Р. Люксембург, 110 Тел.+375291307140, E- mail: tat_vaikh@mail.ru |
Рисунок 1. Этапы созревания преламина в ламин А
Ламины – это структурные белки, компоненты ядерной ламины – белковой сети, которая лежит под внутренней мембраной ядра и определяет его размер и форму. Ядерная ламина обеспечивает прочность ядерной оболочки и организацию ядерных пор, противостоит силам деформации и защищает хроматин от физических повреждений. Как показывают исследования последних лет, наряду с выполнением структурной функции, ламины принимают участие в контроле репликации ДНК, организации хроматина и в регуляции генной экспрессии, процессинга и апоптоза [1-4].
Ядерная ламина состоит из четырех ламиновых белков: A, B1, B2 и C. Ламины В1, В2 и B3 (их называют ламинами В-типа) кодируются двумя генами, LMNB1 и LMNB2 и синтезируются во всех клетках многоклеточных животных. Ламины А и С (так называемые ламины А-типа) являются продуктами альтернативного сплайсинга одного гена LMNA и обнаруживаются в сравнимых количествах в дифференцированных тканях всех позвоночных, в т.ч. человека. Все белки, ламины типа В и преламин А, синтезируются с консервативным мотивом на карбоксильном конце молекулы – СааХ (С – цистеин, аа – две алифатические аминокислоты, Х – любая аминокислота) [5,6].
Преламины А и В для превращения в ламины (рис. 1: А-Д) подвергаются целой серии посттрансляционных модификаций, которые происходят на карбоксильном конце молекулы: А) фарнезилирование цистеина из CaaX мотива; Б) эндопротеолиз и высвобождение последних трех аминокислот белка (ааХ); С) метилирование вновь образованного фарнезилци-стеина; Д) второй эндопротеолиз, отщепляющий дополнительно 15 аминокислот с карбоксильного конца, в том числе и фарнезилцистеин.
У ламинов В второго протеолиза нет, они остаются с фарне-зильной группой на С-конце. Функцией фарнезильной группы является установление связи ламина с ядерной мембраной либо путем прямого взаимодействия с липидной мембраной, либо опосредованно, с помощью белок-белковых взаимодействий [7,8].
Молекулы ламинов имеют массу 60–89 кДа и трехчленную структуру, состоящую из центрального альфа-спирального
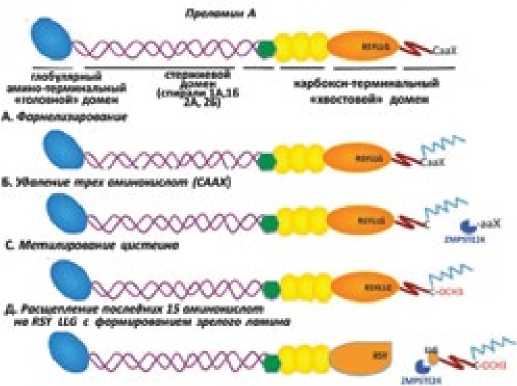
Примечание: RSYLLG – аминокислотные остатки на С-конце белка в последовательности, указанной в однобуквенной номенклатуре (Ali J. Marian, Сardiogenetics, 2011, 1e6, с изменениями).
стержня, ограниченного коротким глобулярным амино-терминальным «головным» доменом и длинным карбокси-терминальным «хвостовым» доменом. Большая часть ламиновых белков сконцентрирована в ядерной ламине, однако небольшая фракция обычно обнаруживается в нуклеоплазме [9]. Гены, кодирующие ламиновые белки, представлены в таблице 1.
Отдельные ламиновые белки А и В типа полимеризуются в клетках в гомополимеры, которые могут объединяться в ламине в единую сетчатую структуру, тогда как в нуклеоплазме такой упорядоченности не обнаруживается. Экспериментально, при последовательном избирательном сайленсинге каждого из ламиновых генов доказано, что формирование единой ламиновой сети происходит под контролем гена LMNB1. Выключение LMNA или LMNB2, в отличие от LMNB1, не оказывало влияния на структуру сети ламиновых белков в целом.
Таблица 1. Гены ламинов и кодируемые ими белки
|
Ген |
Локус на хромосоме |
Число экзонов |
Белки, количество аминокислот |
Примечания (тип синтеза) |
|
LMNA |
1q22 |
17 |
Ламин А - 654 Ламин С - 572 |
Альтернативный сплайсинг LMNA |
|
LMNB1 |
5q23.2 |
12 |
Ламин B1 - 586 |
Обычный |
|
LMNB2 |
19p13.3 |
14 |
Ламин B2 – 620 Ламин В3 – 483 |
Альтернативный сплайсинг |
Предполагается, что ламиновый ген LMNB1 контролирует образование структурной основы ядерной ламины. Выявлено, что ламиновые белки B1 и B2 собираются в отдельные, но взаимосвязанные сети и по-разному взаимодействуют с сетью, образованной ламинами А и С. Установлено также, что ламиновые белки А-типа преимущественно ассоциируются с участками хроматина, богатыми генами [10], тогда как участки хромосом с малым числом генов связаны в ламине с белками B1 [11]. В нормальных клетках взаимодействие ламинов А- и В-типов между собой и с хроматином ядра создает возможность тонкой регуляции экспрессии генов. Нарушение этого процесса при поражении любого из ламиновых компонентов лежит в основе так называемых ламинопатий [10].
Ламинопатии. Заболевания, развивающиеся в результате мутаций в генах белков ядерной ламины, называют лами-нопатиями. При этом, вышеуказанные синдромы могут вызываться мутациями как в генах собственно ламинов, так и в генах белков-партнеров (SREBP1, эмерины) и ферментов, участвующих в процессинге ламинов [12]. Клинические синдромы ламинопатий были описаны еще в конце 19-го века, однако лишь в 1999 году мутация гена ламина А/С была впервые выявлена у пациентов с аутосомно-доминантным заболеванием – мышечной дистрофией Эмери-Дрейфуса (EDMD) [13,14]. Позже с ламиновыми мутациями были ассоциированы более десятка клинических расстройств, формирующих уже известные синдромы с поражением, преимущественно, определенного вида ткани: поперечно-полосатые мышцы, жировая ткань, сердечный миокард, периферические нервы или множественные ткани в результате прогероидных фенотипов [15]. В таблице 2 приведены известные к настоящему времени синдромы и заболевания, генетически детерминированные ламиновыми мутациями.
Точный механизм развития ламин-ассоциированных заболеваний до сих пор еще детально не изучен. Доминируют две основные гипотезы, объясняющие наблюдаемые патологические фенотипы: структурная гипотеза и гипотеза «генной экспрессии». Согласно первой, недостаток ламинов или некорректная сборка мутантных ламиновых белков приводит к снижению прочности ядерной ламины и повышению уязвимости ядра и клетки в целом [1,16]. Прежде всего при этом страдают клетки, подвергающиеся механическому стрессу, такие как мышечные клетки и кардиомиоциты, с развитием дегенеративных изменений [17]. Вторая гипотеза предполагает нарушение взаимосвязи между ядерной ламиной и факторами транскрипции [2,18,19]. Недавно была сформулирована еще одна гипотеза, согласно которой мутация ламинов A/C или отсутствие ламинов А-типа могут провоцировать третий механизм патогенеза – временную декомпартментализацию (из-за нарушения целостности ядерной мембраны), приводящую к неадекватному обмену между ядерными и цитоплазматическими компонентами [20].
Кардиомиопатии, вызванные генными мутациями LMNA, часто сопровождаются скелетно-мышечными расстройствами (периферические миопатии) различной степени выраженности. Чаще встречается мышечная дистрофия Эмери-Дрейфуса аутосомно-доминантного типа и поясно-конечностная мышечная дистрофия с нарушением атриовентрикулярной проводимости типа 1В. Иногда отмечаются изолированные скелетно-мышечные аномалии в виде мускульных гипо/ги-пертрофий конечностей.
Предварительный диагноз ламин-ассоциированной ДКМП можно предположить у пациентов с дефектами проводимости (атриовентрикулярная блокада или хронотропная дисфункция синусового узла); при сочетании ДКМП с аномалиями скелетных мышц (мышечная слабость/миопатии, сухожильные контрактуры, повышение уровня креатинфосфокиназы); при появлении суправентрикулярной или желудочковой тахиаритмии через несколько лет после выявления ДКМП. Дефекты проводимости, желудочковые и наджелудочковые аритмии манифестируют в возрасте 20-30 лет. Желудочковые тахиаритмии могут возникать на различных стадиях заболевания. Миопатический комплекс (слабость/миопатия скелетных мышц) может отсутствовать или присоединиться в поздней стадии болезни. Хронология симптомов и клинические проявления заболевания могут различаться между родственниками внутри одной семьи.
В качестве примера представляем два клинических случая ламин-ассоциированной ДКМП с сопутствующим миопатическим синдромом различной степени выраженности: первое наблюдение заболевания (пациентка К.) с быстрым прогрессирующим течением кардиомиопатии, бивентрикулярной дисфункцией и развитием тяжелой сердечной недостаточности (СН), сопровождающееся легкой изолированной скелетно-мышечной аномалией в виде умеренной гипотрофии бедренных мышц; второе наблюдение (пациент С.) с медленно-прогрессирующим течением ДКМП, умеренно выраженной СН и прогрессирующей мышечной дистрофией Эмери-Дрейфуса с тяжелыми нарушениями опорно-двигательных функций. Общей в двух случаях была кардиальная манифестация заболевания в виде аритмических и синкопальных эпизодов; и в первом, и во втором случае выявлены идентичные (синдром Фредерика) нарушения ритма (фибрилляция предсердий) и проводимости (атриовентрикулярная блокада 3 степени) с синкопе.
Клинический случай 1. Пациентка К., женщина 1991 г.р., без семейного анамнеза ДКМП. Первые жалобы в виде чувства сердцебиения и нарушения сердечного ритма появились в 21-летнем возрасте. При электрокардиографическом (ЭКГ) обследовании выявлены: низкий вольтаж комплексов QRS в стандартных и грудных отведениях, плохо визуализируемый низкоамплитудный зубец Р, синусовая брадикардия с ЧСС 50 уд. в 1мин, АВ блокада 1 ст., одиночные предсердные экстрасистолы, в т.ч. с блокированным проведением. При ультразвуковом исследовании сердца морфо-функциональной патологии не обнаружено: конечно-диастолический диаметр левого
Таблица 2. Клинические синдромы кардиальных и экстракардиальных ламинопатий
|
Ламинопатии |
Клинические проявления |
Тип наследования |
|
Мышечная дистрофия Эмери-Дрейфуса 2 (EDMD2) |
Ранние контрактуры локтей, ахиллова сухожилия и задней части шеи, жесткость позвоночника, медленно прогрессирующая мышечная слабость в руках и голенях, дилатационная кардиомиопатия с АВ блокадой |
Аутосомно-доминантный |
|
Мышечная дистрофия Эмери-Дрейфуса 3 (EDMD3) |
Общая схожесть с EDMD2. Контрактуры, диффузная мышечная дистрофия, отсутствие кардиомиопатии |
Аутосомно-рецессивный |
|
Дилатационная кардиомиопатия 1А (CMD1A) |
Дилатация и дисфункция миокарда, нарушения ритма и проводимости (АВ блокада и ПБЛНПГ), мышечная слабость, мышечная дистрофия, ограничение подвижности в суставах, контрактуры |
Аутосомно-доминантный |
|
Конечностно-поясная мышечная дистрофия 1В (LGMD1В) |
Прогрессирующая проксимальная мышечная слабость и гипотрофии, симптомы «крыловидных лопаток», «утиной походки», поясничный гиперлордоз |
Аутосомно-доминантный |
|
Частичная семейная липодистрофия Даннигана (FPLD2) |
Пациенты рождаются с нормальным распределением жира, но после наступления половой зрелости проявляются региональные потери жира конечностей, сопутствующие сахарный диабет или резистентность к инсулину |
Аутосомно-доминантный |
|
Болезнь Шарко-Мари-Тута 2 типа (CMT2B1) |
Характерно снижение нервной чувствительности, потеря больших миелинизированных волокон и аксональная дегенерация |
Аутосомно-рецессивный |
|
Мандибуло-акральная дисплазия тип А (МАDА) |
Характерна задержка роста,пациенты имеют уменьшенную челюсть, недоразвитые ключицы, другие врожденные аномалии скелета, частичную липодистрофию и прогероидные признаки |
Аутосомно-рецессивный |
|
Прогерия Хатчинсона- Гилфорда (HGPS) |
Проявляются признаки ускоренного или преждевременного старения. Обычно пациенты умирают на втором десятилетии жизни от инфаркта миокарда или инсульта. Другими признаками являются склеротические изменения кожи, контрактуры суставов, выпуклые глаза, уменьшенные челюсти, уменьшение подкожно-жировой клетчатки, выпадение волос, пятнистость кожи и видимая подкожная сосудистая сеть, нарушения роста |
Аутосомно-доминантный |
|
Рестриктивная дермопатия |
Задержка роста, плотная ижесткая кожа, с эрозией на месте сгибов, поверхностные сосуды, облысение, микрогнатия и другие нарушения костей |
Аутосомно-рецессивный |
|
Синдром рука-сердце, словенский тип |
Сочетание различны Сочетание различных пороков развития верхних конечностей и врожденных пороков сердца. Степень мальформации рук различна. Кроме этого, характерны другие нарушения скелета. Наблюдаются дефекты межпредсердной и межжелудочковой перегородок, открытый артериальный проток, коарктация аорты, стеноз легочной артерии, пролапс митрального клапана |
Аутосомно-доминантный |
желудочка (КДД ЛЖ) 48 мм, конечно-систолический диаметр (КСД) ЛЖ 29 мм, конечно-диастолический объем (КДО) ЛЖ 123 мл, конечно-систолический объем (КСО) ЛЖ 47 мл; конечно-диастолический объем правого желудочка (КДО ПЖ) 63 мл, конечно-систолический объем ПЖ 28 мл, фракция выброса (ФВ) ЛЖ 62% (по Симпсону), фракция выброса правого желудочка (ФВ ПЖ) 55%. Через два года пациентка госпитализирована в республиканский кардиологический центр с клиническими признаками сердечной недостаточности (СН), соответствующими III ФК NYHA, с жалобами на приступы удушья по ночам, тяжесть и боли в правой подреберной области, одышку, синкопе и предобморочные состояния. Клиника СН развивалась быстро, в течение 3-х месяцев с момента появления первых симптомов – одышки и слабости.
Объективно при осмотре: ЧДД 23 в 1 мин, PS = 51, ЧCC = 60 уд. в 1 мин., АД 100/60 мм рт. ст.; перкуторно – расширение границ сердца, разлитой верхушечный толчок; аускультатив-но – сердечные тоны аритмичные, систолический шум над мечевидным отростком, мелкопузырчатые хрипы в нижних отделах легких; пальпаторно – увеличение правой доли печени (+2,5 см от края реберной дуги); локально – периферические отеки стоп и голеней, признаки гипертрофии икроножных мышц, умеренно выраженная гипотрофия бедренных мышц (преимущественно квадрицепсы) без снижения мышечной силы и тонуса с нормальными периостальными рефлексами. На ЭКГ: фибрилляция предсердий с ЧСС 51-60 в 1 мин, неполная блокада левой ножки пучка Гиса (альтернирующие блокады передневерхнего и задненижнего разветвления), интермиттирующая полная блокада правой ножки пучка Гиса. При эхокардиографическом (Эхо-КГ) исследовании выявлена дилатация и глобальная систолическая дисфункция обоих желудочков с дилатацией предсердий: ФВ ЛЖ 27%, среднее значение продольной деформации ЛЖ (mean global strain – ср.GS) – 8,6%; ФВ ПЖ 39%, продольная деформация ПЖ (ср. GS) – 9,8%; КДД ЛЖ 56 мм (индекс 28 мм/м2), КСД ЛЖ 46 мм, КДО ЛЖ 171 мл, КСО ЛЖ 124 мл; КДО ПЖ 122 мл, КСО ПЖ 74 мл; левопредсердный объем составил 78 мл, объем правого предсердия – 200 мл; среднее давление в легочной артерии (ср.ДЛА) – 28 мм рт. ст. Обнаружены признаки трабекулярного строения верхушки ЛЖ, заднебоковых отделов ЛЖ, верхушки ПЖ, межжелудочковой перегородки со стороны ПЖ. С помощью кардиореспираторного теста выявлено пиковое потребление кислорода (VO2) в последние 30 сек физической нагрузки – пик VO2 – 15,5 мл/кг/мин (диапазон половозрастной нормы 29-42 мл/кг/мин). При суточном мониторировании ЭКГ выявлены следующие нарушения сердечного ритма и проводимости: фибрилляция предсердий (ФП) брадисисто-лической формы со средней ЧСС 45 уд. в 1 мин; частая полиморфная желудочковая экстрасистолия (>300/час), в т.ч. парная (345 куплетов/сут) и групповая (13 триплетов/сут); в ночное и утреннее время зарегистрированы альтернирующие эпизоды синдрома Фредерика (АВ блокада 3 степени дистального типа общей длительностью 15 час/сут с минимальной ЧСС 24 уд. в 1 мин) и 40 пароксизмов неустойчивой желудочковой тахикардии двух морфологических форм (в виде блокады правой ножки пучка Гиса и задненижнего разветвления левой ножки пучка Гиса).
При лабораторно-биохимическом исследовании крови выявлено повышение уровней сывороточной креатинфосфокиназы – 293 U/L (норма 24-190 U/L) и мозгового натрийуретического пептида – 789 пг/ммоль (норма 0-50 пг/ммоль). При селективной ангиографии коронарных артерий видимых патологических изменений коронарного русла не обнаружено. С помощью сцинтиграфии миокарда (в состоянии покоя) выявлены зоны гипоперфузии в передней, переднебоковой, передневерхушечной и задней областях ЛЖ.
Магнитно-резонансное исследование выполнено пациентке на абдоминальной катушке в неполном объеме из-за значительных клинических проявлений левожелудочковой СН (приступы ОЛЖН) в горизонтальном положении в условиях сканирования. Выявлены признаки диффузной дилатации всех камер сердца; расширение легочного ствола до 44 мм (превышение диаметра аорты); снижение глобальной сократимости миокарда ЛЖ и ПЖ без признаков истончения и без жировой инфильтрации миокарда и эпикарда; повышенная трабекулярность миокарда ПЖ; жидкость в полости перикарда и в видимых отделах брюшной полости.
С целью первичной профилактики ВС пациентке проведена имплантация однокамерного кардиовертера-дефибриллятора (КВД).
Несмотря на проведение адекватной медикаментозной терапии (базовое лечение СН ингибиторами АПФ, бета-бло-

каторами, антагонистами альдостерона, диуретиками), наблюдалось дальнейшее прогрессирование симптомов СН с развитием асцита и гидроперикарда. Пациентку с клиническими признаками сердечной недостаточности, резистентной к фармакотерапии, и прогрессирующим негативным ремоделированием сердца (увеличение КДО ЛЖ до 180 мл и КСО ЛЖ до 140 мл; снижение ФВ ЛЖ до 22% и ср.GS ЛЖ до – 5,2%; увеличение КСО ПЖ до 93 мл и КДО ПЖ до 131 мл; уменьшение ФВ ПЖ до 29% и ср. GS ПЖ до – 4,9%; появление признаков выраженной легочной гипертензии со ср. ДЛА = 46 мм рт. ст.) включили в лист ожидания пересадки сердца с последующей дальнейшей успешной ортотопической трансплантацией сердца.
С согласия пациентки проведено молекулярно-генетическое исследование гена LMNA методом анализа конформационного полиморфизма одноцепочечных фрагментов ДНК (SSCP) и прямого секвенирования. В экзоне III гена LMNA было выявлено гетерозиготное носительство нуклеотидной замены G>C (рис. 2).
Рисунок 2. Результаты молекулярно-генетического исследования гена LMNA у пациентки К
Мутация Мутация
Список литературы Мутации гена ламина A/C (LMNA) у пациентов с дилатационной кардиомиопатией и их фенотипические проявления
- Burke B, Stewart CL. Life at the edge: the nuclear envelope and human disease. Nat Rev Mol Cell Biol 2002; 3:575-585
- Cohen M, Lee KK, Wilson KL, Gruenbaum Y. Transcriptional repression, apoptosis, human disease and the functional evolution of the nuclear lamina. Trends Biochem Sci 2001; 26:41-47
- Lammerding J, Schulze PC, Takahashi T et al. Lamin A/C deficiency causes defective nuclear mechanics and mechanotransduction. J Clin Invest 2004;113(3): 370-8
- Hutchison CJ. Lamins: building blocks or regulators of gene expression? Nat Rev Mol Cell Biol 2002; 3(11): 848-58
- Lin F, Worman HJ. Structural organization of the human gene encoding nuclear lamin A and nuclear lamin C. J Biol Chem 1993; 268:16321-16326
- Dechat T. Nuclear lamins: Major factors in the structural organization and function of the nucleus and chromatin. Genes Dev 2008; 22: 832-853.
- Mattout A, Dechat T, Adam S A, Goldman and Gruenbaum Y. Nuclear lamins, diseases and aging//Current Opinion in Cell Biology 2006,18:335-341
- Meshorer E, Gruenbaum Y. Gone with the Wnt/Notch: stem cells in laminopathies, progeria, and aging. J Cell Biol 2008; 181(1): 9-13
- Moir R., Yoon M., Khuon S., Goldman R. Nuclear lamins A and B1: Different pathways of assembly during nuclear envelope formation in living cells. J Cell Biol 2000;151:1155-1168
- Shimi T, Pfleghaar K, Kojima S, et al. The A-and B-type nuclear lamin networks: microdomains involved in chromatin organization and transcription//Genes Dev 2008; 22(24): 3409-3421
- Guelen, L., Pagie, L., Brasset, E., et al. Domain organization of human chromosomes revealed by mapping of nuclear lamina interactions. Nature 2008; 453:948-951
- Rajendran V., Purohit R., Sethumadhavan R. In silico investigation of molecular mechanism of laminopathy caused by a point mutation (R482W) in lamin A/C protein. Amino Acids 2012; 43 (2): 603-615
- Bonne G., Di Barletta M.R., Varnous S. et al. Mutations in the gene encoding lamin A/C cause autosomal dominant Emery-Dreifuss muscular dystrophy. Nat Genet 1999; 21:285-288
- Fatkin D., MacRae C., Sasaki T. et al. Missense mutations in the rod domain of the lamin A/C gene as causes of dilated cardiomyopathy and conduction-system disease. N Engl J Med 1999; 341:1715-1724
- Worman H., Ostlund C., Wang Y. Diseases of the nuclear envelope. Cold Spring Harb Perspect. Biol 2010; 2:760-776
- Houben F. De Vos W. H. Krapels I. P. et al. Cytoplasmic localization of PML particles in laminopathies. Histochem Cell Biol 2013; 139:119-134
- Hutchison CJ, Alvarez-Reyes M, Vaughan OA. Lamins in disease: why do ubiquitously expressed nuclear envelope proteins give rise to tissue-specific disease phenotypes? J Cell Sci 2001; 114(1): 9-19
- Wilson KL, Zastrow MS, Lee KK. Lamins and disease: insights into nuclear infrastructure. Cell 2001; 104(5): 647-650
- Schirmer EC, Foisner R. Proteins that associate with lamins: many faces, many functions. Exp Cell Res 2007; 313(10): 2167-2179
- De Vos WH, Houben F, Kamps MV et al. Repetitive disruptions of the nuclear envelope invoke temporary loss of cellular compartmentalization in laminopathies. Hum Mol Genet 2011; 20:4175-4186
- Dhe-Paganon S1, Werner ED, Chi YI, Shoelson SE. J Structure of the globular tail of nuclear lamin. Biol Chem 2002; 277(20): 17381-17384.
- Verdaguer N, Corbalan-Garcia S, Ochoa WF Fita et al. Ca(2+) bridges the C2 membrane-binding domain of protein kinase Calpha directly to phosphatidylserine. J EMBO 1999; 18(22): 6329-6338
- Hershberger RE, Morales A, Siegfried JD. Clinical and genetic issues in dilated cardiomyopathy: A review for genetics professionals. Genet Med 2010; 12:655-667
- Van Tintelen J. Peter, Petronella G. Pieper, Karin Y. Van Spaendonck-Zwarts et al. Pregnancy, cardiomyopathies, and genetics. Cardiovascular Research 2014; 101:571-578
- Jacoby D, McKenna WJ. Genetics of inherited cardiomyopathy. Eur Heart J 2012; 33:296-304
- Fatkin D, Otway R, Richmond Z. Genetics of dilated cardiomyopathy. Heart Fail Clin 2010; 6:129-140
- R.Weichenhan, T.M.Strom, A.Pfeufer et al. Cardiomyopathies. Targeted Next-Generation Sequencing for the Molecular Genetic Diagnostics of Cardiomiopathies. J Circ Cardiovasc Genet 2011; 4:110-122
- Van Berlo JH, Meune C, Anselme F et al. Meta-analysis of clinical characteristics of 299 carriers of LMNA gene mutations: do lamin A/C mutations port end a high risk of sudden death? J Mol Med 2005; 83:79-83
- Pasotti M, Klersy C, Pilotto A, et al. Long-term outcome and risk stratification in dilated cardiolaminopathies. J Am Coll Cardiol 2008; 52:1250-1260
- Ph.Charron, E. Arbustini, G. Bonne. What Should the Cardiologist know about Lamin Disease? Arrhythmia & Electrophysiology 2012; 1:22-28
- Parks SB, Kushner JD, Nauman D et al. Lamin A/C mutation analysis in a cohort of 324 unrelated patients with idiopathic or familial dilated cardiomyopathy. A. Heart J 2008; 56:161169
- Meune C, Van Berlo JH, Anselme F et al. Primary prevention of sudden death in patients with lamin A/C gene mutations. N Engl J Med 2006; 354:209-210
- Fernandez X, Dumont CA, Monserrat L et al. Sudden death in a patient with lamin A/C gene mutation and near normal left ventricular systolic function. Int J Cardiol 2008; 126: 136137
- Ehlermann P, Lehrke S, Papavassiliu T, et al. Sudden cardiac death in a patient with lamin A/C mutation in the absence of dilated cardiomyopathy or conduction disease. Clin Res Cardiol 2011; 100:547-551
- Van Rijsingen IA, Arbustini E, Elliott PM, et al. Risk factors for malignant ventricular arrhythmias in lamin A/C mutation carriers a European cohort study. J Am Coll Cardiol 2012; 59: 493-500
- Charron P, Arad M, Arbustini E, et al. Genetic counselling and testing in cardiomyopathies: a position statement of the European Society of Cardiology Working Group on Myocardial and Pericardial Diseases. Eur Heart J 2010; 1:2715-2726
- Frey N, Katus HA. Dilated cardiomyopathy as a genetic disease: molecular and clinical aspects. Internist 2008: 49: 43-50
- Hershberger RE, Morales A, Siegfried JD. Clinical and genetic issues in dilated cardiomyopathy: A review for genetics professionals. Genet. Med 2010; 12:655-667
- Jacoby D, McKenna WJ. Genetics of inherited cardiomyopathy. Eur. Heart J 2012; 33:296-304
- Fatkin D, Otway R, Richmond Z. Genetics of dilated cardiomyopathy. Heart Fail. Clin 2010; 6:129-140
- Hugh Watkins, Houman Ashrafian, D.Phil., Charles Fedwood. Inherited Cardiomyopathies. New Engl. J Medicine 2011; 364(17): 1643-1656
- Hershberger RE, Siegfried JD. Update 2011: clinical and genetic issues in familial dilated cardiomyopathy. J Am. Coll. Cardiol 2011; 57:1641-1649
- Вайханская Т.Г., Сивицкая Л.Н, Даниленко Н.Г. и соавт. Что должен знать сегодня кардиолог о дилатационной кардиомиопатии, связанной с мутацией гена ламина (LMNA)? Кардиология в Беларуси 2013; 4 (29): 64-81.
