Наследственная увеальная меланома: обзор литературы и клинический случай

Автор: Семьянихина Александра Владимировна, Филиппова Маргарита Геннадьевна, Архипова Оксана Николаевна, Любченко Людмила Николаевна
Журнал: Сибирский онкологический журнал @siboncoj
Рубрика: Обзоры
Статья в выпуске: 2 т.17, 2018 года.
Бесплатный доступ
Увеальная меланома (УМ) - самое частое первичное злокачественное новообразование увеального тракта с молекулярно-генетическими характеристиками, позволяющими отличать ее от других подтипов меланомы. Соматические мутации в опухоли при УМ вовлекают ряд генов - BAP1, EIF1AX, GNA11, GNAQ и SF3B1, которые определяют биологию и поведение опухоли, являясь предикторами течения заболевания. Примерно в 2-5 % случаев развитие УМ ассоциировано с наследственной патологией и связано с герминальными мутациями в генах, ответственных за тот или иной синдром. На данный момент описано несколько таких синдромов, среди которых BAP1 -ассоциированный синдром, FAMMM-синдром, синдром Ли-Фраумени и др. В настоящей статье приводится анализ современного представления о природе наследственной УМ и рассматривается клинический случай семейной УМ.
Увеальная меланома, наследственные синдромы, ген chek2, ли-фраумени-подобный синдром
Короткий адрес: https://sciup.org/140254179
IDR: 140254179 | УДК: 616-006.81 | DOI: 10.21294/1814-4861-2018-17-2-82-88
Hereditary uveal melanoma: a review of literature and a case report
Uveal melanoma (UM) is the most common primary intra-ocular malignancy. Uveal melanoma is distinct from other subtypes of melanoma by its molecular and genetic characteristics. Somatic mutations in UM tumor involve genes, such as BAP1, EIF1AX, GNA11, GNAQ and SF3B1, that determine the biology and behavior of a tumor and appear to be predictors of disease. In 25 % of cases, the development of UM is associated with hereditary diseases and can be caused by germline mutations in genes that are responsible for a particular syndrome. Several such syndromes ( BAP1 -associated syndrome, FAMMM-syndrome, Li-Fraumeni syndrome and etc.) have been identified. In this article we analyze the modern concept of the nature of hereditary UM and present the case of hereditary UM.
Текст научной статьи Наследственная увеальная меланома: обзор литературы и клинический случай
Увеальная меланома (УМ) является вторым самым частым подтипом меланомы (на ее долю приходится около 5 %) и самой распространен‑ ной первичной интраокулярной злокачественной опухолью, поражающей один из отделов увеаль‑ ного тракта, включая радужку, цилиарное тело и сетчатку [1]. В 2–3 % случаев УМ локализуется в конъюнктиве глаза [2]. Ежегодная заболеваемость УМ колеблется от 5 до 14 случаев на 1 млн. Сред‑ ний возраст манифестации опухоли составляет 60 лет. Незначительно чаще УМ встречается у мужчин [3]. В группу повышенного риска развития УМ относят обладателей светлых кожи и волос, голу‑ бых глаз, веснушек и невусов [1]. Представители европеоидной расы болеют УВ примерно в 150 раз чаще. Несмотря на успехи последних лет в лечении первичной УМ, пятилетняя выживаемость состав‑ ляет 72–84 %, а у половины пациентов диагности‑ руется прогрессирование основного заболевания в первые декады после начала специфического лечения. Более чем в 90 % случаев фатальное те‑ чение заболевания ассоциировано с гематогенным метастазированием в печень [3].
Клинические и молекулярно‑биологические характеристики УМ, несмотря на общность гисто‑ генеза, значительно отличают ее от других подти‑ пов меланомы (меланомы кожи (МК) и слизистых оболочек). Экзогенные факторы, повышающие риск развития МК, такие как чрезмерная инсоля‑ ция, не нашли достоверного подтверждения для УМ, а вклад ультрафиолетового излучения в ини‑ циацию канцерогенеза при УМ до сих неясен [4]. При УМ гораздо реже встречаются типичные для МК молекулярно‑генетические перестройки, во‑ влекающие гены RAS‑каскада (BRAF, NRAS, NF1). Применение современных ДНК‑диагностических методов, в том числе секвенирования нового по‑ коления, позволило выявить несколько драйверных генов с высокой частотой соматических мутаций в образцах УМ [2]. К ним относятся гены BAP1, EIF1AX, GNA11, GNAQ и SF3B1. В большинстве случаев мутации в этих генах взаимно исключают друг друга, несут различный вклад в риск мета‑ стазирования и имеют разную прогностическую значимость (табл. 1).
Подавляющее большинство случаев УМ явля‑ ются спорадическими, т.е. обусловленными раз‑ личными мутациями в клетке‑предшественнике меланоцитарного звена, дающего начало патологи‑ ческому опухолевому клону. Однако еще в 1892 г. английским ученым Silcock было высказано пред‑ положение о возможной наследственной причине в ряде случаев УВ. Поводом послужило наблюдение семьи с накоплением случаев УМ и рака молоч‑ ной железы (РМЖ) в четырех поколениях [8, 9]. С момента данной публикации более 50 научных
таблица 1
Основные молекулярно-генетические различия между УМ и МК [5, 6, 7]
Показатель Меланома кожи Увеальная меланома
Уровень геномной нестабильности Высокий Низкий
Хромосомные аберрации Часто Редко
Хромосомные аберрации 1р‑, 1q+, 6p+, 6q‑, 8p‑, 8q+, 11q‑ , 1р‑, 1q+, 6p+, 6q‑, 8p‑, 8q+, 11q‑ , моносомия 3 (редко), 9р‑ и 10р (часто) моносомия 3 (часто), 9р‑ и 10р (редко)
mutBRAF/NRAS + +/ mutGNAQ/ GNA11
mutBAP1 +/ работ было посвящено семейной форме УМ [1, 2, 10–12]. В подавляющем большинстве случаев и до недавнего времени значительная часть авторов склонялась к полигенной природе УМ [2, 13, 14]. Однако с расширением возможностей современной ДНК‑диагностики на протяжении последних лет активно изучается роль наследственного фактора в развитии УМ и проводится поиск вероятных генов‑кандидатов.
Несмотря на то, что большая часть описанных случаев семейной УМ не была ассоциирована с синдромальной патологией, частота УМ возрастает среди ряда наследственных синдромов. В 1937 г. появляется первая публикация, посвященная описанию клинического наблюдения – манифе‑ стации УМ у пациента с нейрофиброматозом (болезнью Реклингхаузена) [8, 15]. Кумулятивный анализ последующих немногочисленных научных работ показал доминирование женского пола и периферического типа нейрофиброматоза в слу‑ чае диагностирования УМ у больных с болезнью Реклингхаузена.
Другим факоматозом, при котором возрастает риск развития УМ, является синдром Горлина (син‑ дром базально‑клеточных невусов), или «пятый фа‑ коматоз», симптомокомплекс, характеризующийся поражением кожи, мышц, скелета и глаз, а также повышенным риском развития злокачественных новообразований (ЗН), прежде всего, кожи и цен‑ тральной нервной системы. Впервые описанная Kedem et al. в 1970 г. УМ в составе синдрома явля‑ ется крайне редким его проявлением [8, 16].
Возрастает частота УМ при наследственных формах МК и слизистых оболочек. Описаны случаи УМ в составе синдрома множественных атипичных невусов и меланомы (так называе‑ мый FAMMM‑синдром, familial atypical mole and melanoma syndrome) [17], фенотипического про‑ явления герминальных мутаций в гене CDKN2A, ответственном за 20–40 % семейных форм МК и около 1 % наследственно обусловленной УМ [17, 18]. В 1000 раз возрастает риск развития УМ у пациентов с другим редким и тяжелым наслед‑ ственным заболеванием, поражающим покровный эпителий, – пигментной ксеродермой [19].
+ +
Ряд исследований, посвященных сочетанию УМ с РМЖ, позволил рассматривать ген BRCA2 как один из генов‑кандидатов, герминальные му‑ тации в котором определяют повышенный риск развития УМ [1]. В среднем частота наследуемых патогенных вариантов в гене BRCA2, ответствен‑ ном за синдром наследственного рака молочной железы и рака яичников, в различных выборках с семейным накоплением УВ и РМЖ составляет от 2 до 5 % [20].
Шведскими учеными при анализе трех семей с множественными случаями УМ и МК и отсут‑ ствием наследуемых мутаций в генах CDKN2A, CDK4, BRCA1 и BRCA2 был выделен локус 9q21.32 на длинном плече 9 хромосомы. Однако попытки картировать гены‑кандидаты, расположенные в данном регионе, не увенчались успехом [21].
Одним из признаков наследственных форм ЗН является наличие у пациента первично‑ множественных злокачественных новообразований (ПМЗН). Сочетание УМ с другими ЗН, в частности с мезотелиомой, стало предметно изучаться с 1970 г., когда появились первые публикации [22]. Часть таких работ посвящена случаям злокаче‑ ственной мезотелиомы с семейным накоплением различных ЗН. Молекулярно‑диагностический поиск в таких семьях позволил выделить новый наследственный синдром, обусловленный герми‑ нальными мутациями в гене BAP1.
BAP1- ассоциированный наследственный син‑ дром, на долю которого приходится 3–4 % от всех случаев УМ [22], характеризуется специфическими кожными проявлениями – атипичными невусами Шпица и повышенным риском развития ряда ЗН: УМ, злокачественной мезотелиомы, МК, светло‑ клеточного рака почки и базально‑клеточного рака, а также ПМЗН. Частота проявления заболевания у носителей герминальных мутаций в гене BAP1 стремится к 85 %, по данным разных авторов, и позволяет отнести данный синдром в группу высокопенетрантных состояний [22–24]. Как и для большинства наследственно обусловленных заболеваний, для BAP1- ассоциированного наслед‑ ственного синдрома характерен более молодой средний возраст манифестации ЗН по сравнению с
|
Наследственные синдромы, ассоциированные с УМ |
таблица 2 |
|||
|
Синдром |
Ген |
Тип наследования Частота |
Другие ЗН, в составе синдрома |
|
9q21.32
общей популяцией. УМ, являясь самой частой опу‑ холью в составе синдрома, встречается примерно в 30 % случаев, имеет более агрессивное течение с высоким риском метастазирования, что опреде‑ ляет худшую выживаемость таких пациентов [22]. Специфического лечения для данного синдрома на сегодняшний день не разработано в связи с от‑ носительно «молодым» возрастом его открытия. Наблюдение за пациентами включает в себя еже‑ годный расширенный осмотр офтальмоонкологом начиная с 11‑летнего возраста, консультации дер‑ матолога и ежегодное УЗИ почек и забрюшинного пространства с 20 лет. Скрининговых мероприятий для своевременной диагностики злокачественной мезотелиомы нет, в качестве альтернативы может быть рассмотрено МРТ брюшной полости с оцен‑ кой состояния брюшины и плевры, если данное ис‑ следование назначено онкологом для исключения специфического поражения почек [23, 24].
Наследственно обусловленная УМ может яв‑ ляться составной частью других редких, феноти‑ пически и генетически гетерогенных, синдрома Ли–Фраумени (СЛФ) и Ли–Фраумени‑подобного синдрома (ЛФПС) [25]. Вслед за описанием гер‑ минальных мутаций в гене TP53 как основного этиологического фактора СЛФ и ЛФПС [26] в начале 1990‑х годов английские офтальмопато‑ логи Jay и McCartney провели ретроспективный генеалогический анализ семьи, впервые описан‑ ной Silcock et al. в 1892 г., с прослеженностью от начала XIX века [27]. Для оценки статуса гена TP53 было выполнено иммуногистохимическое исследование (ИГХ) доступных образцов УМ по‑ раженных членов семьи с целью оценки его экс‑ прессии. Авторам удалось проследить 7 случаев
УМ в сочетании, по меньшей мере, в двух случаях с билатеральным РМЖ. ИГХ‑исследование по‑ казало наличие мутантного типа гена ТР53 в двух пригодных для анализа образцах УМ . Резюмируя полученные результаты, Jay и McCartney предпо‑ ложили, что данная семья может служить одним из ранних примеров синдрома Ли–Фраумени, впервые описанного клиницистами Фредериком Ли и Джозефом Фраумени‑младшим в 1969 г. [25]. Наследуемые мутации в гене ТР53 являются основ‑ ной, но не единственной причиной СЛФ и ЛФПС. Молекулярно‑генетические перестройки в генах CHEK2 и BRCA2 также задействованы в канцеро‑ генезе при данных синдромах, а частота выявления герминальных мутаций в этих генах возрастает при неизмененном статусе гена TP53 . В табл. 2 пред‑ ставлена сводная информация о наследственных синдромах, ассоциированных с УМ.
В зависимости от синдромальной принадлеж‑ ности характерные признаки наследственной УМ могут меняться, при этом остаются такие общие черты:
– составляет 2–5 % от всех вновь диагностиро‑ ванных случаев УМ;
– является полигенным заболеванием со сред‑ ней пенетрантностью (в пораженных семьях встречается не более 2–3 случаев);
– характеризуется более молодым возрастом манифестации заболевания по сравнению с общей популяцией (в среднем 40 лет к общепопуляцион‑ ным 60 годам);
– отмечается тенденция к двустороннему и мультифокальному поражению увеального тракта;
– встречается в составе ПМЗН;
– наличие отягощенного семейного анамнеза у больного УМ (в зависимости от синдромальной патологии наблюдается накопление различных онкологических заболеваний).
Независимо от наследственной природы УМ, по опубликованным данным объединенной рабочей группы по изучению УМ (COMS (Collaborative Ocular Melanoma Study Group)), риск вторых и по‑ следующих опухолей у пациентов с УМ составляет 7,7 % после 5 лет от момента постановки диагноза и 14,9 % – к 10 годам [28]. Самыми частыми ЗН являются: рак предстательной железы у мужчин (в 23 % случаев) и РМЖ у женщин (17 %). Достовер‑ ных данных о повышении риска развития вторых первичных опухолей у пациентов с УМ после про‑ хождения лучевой терапии в плане комплексного лечения не получено [28].
Клинический случай
Пациентка Л., 62 лет, обратилась в научноконсультативное отделение ФГБУ «РОНЦ им. Н.Н. Блохина» Минздрава России в феврале 2016 г. с диагнозом направления: Меланома хориоидеи правого глаза. Состояние после энуклеации правого глаза в 2015 г. Заболевание манифестировало в августе 2015 г. и проявилось полной потерей зрения с пораженной стороны. При обследовании диагностировано злокачественное поражение сетчатки правого глаза – увеальная смешанноклеточная пигментная меланома, узловой тип роста, с инвазией цилиарного тела и частичной отслойкой сетчатки опухолевой тканью. По месту жительства было проведено хирургическое лечение в объеме энуклеации правого глаза.
При контрольном плановом обследовании в ФГБУ «РОНЦ им. Н.Н. Блохина» Минздрава России через 6 мес от момента постановки диагноза выявлено прогрессирование основного заболевания с метастатическим поражением печени. После проведения первой линии химиотерапии (ХТ) паклитакселом в монорежиме отмечался дальнейший рост очагов в печени. Стабилизации процесса удалось добиться после второй линии полихимио-терапевтического лечения (ПХТ) гемцитабином и треосульфаном. По решению консилиума в плане последующего комплексного лечения пациентке в июне 2016 г. выполнена трансартериальная химиоэмболизация (ХЭ) печени (карбоплатин + липиодол). От предложенной повторной ХЭ, а также от продолжения ПХТ больная отказалась. Связь с пациенткой поддерживается. В марте 2017 г. больная жива, самочувствие удовлетворительное, от диагностических мероприятий отказывается.
Учитывая онкологически отягощенный семейный анамнез, больная была направлена на медико-генетическое консультирование (МГК) в лабораторию клинической онкогенетики ФГБУ «РОНЦ им. Н.Н. Блохина» Минздрава России. При сборе семейного анамнеза отмечено, что сестра пациентки, 73 лет, перенесла лечение по поводу увеальной меланомы левого глаза в возрасте 63 лет, а 67-летний брат больной оперирован в связи с раком почки в возрасте 52 лет (рис. 1). Также в семье наблюдались случаи рака поджелудочной железы и женских половых органов по линии отца. Принимая во внимание семейный анамнез, клинико-диагностические данные для исключе-ния/подтверждения наследственной этиологии заболевания на первом этапе было выполнено молекулярно-генетическое исследование с целью поиска герминальных мутаций в генах BRCA1, BRCA2 и CHEK2. В результате ДНК-тестирования в 5 экзоне гена CHEK2 выявлена герминальная миссенс-мутация с.470Т/С (р.Ile157Thr) в гетерозиготном состоянии, зарегистрированная в международной базе данных dbSNP (rs17879961) как клинически значимый патогенный вариант [29].
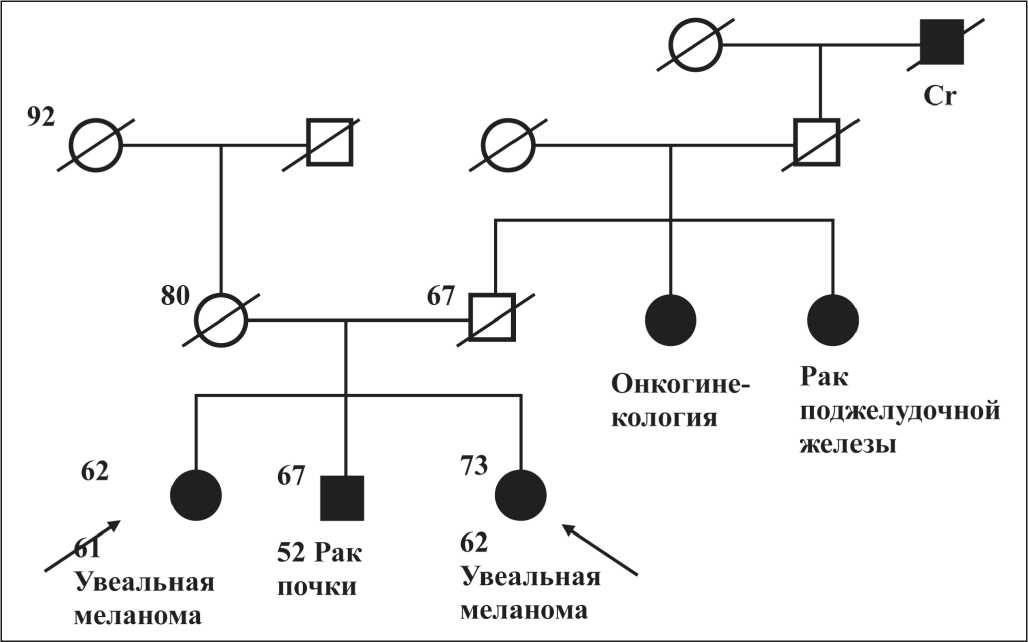
Рис. 1. Родословная пациентки Л.
Обсуждение
Представленное наблюдение является редким клиническим случаем семейной формы УМ и по‑ казательным с точки зрения дифференциального клинико‑генетического диагноза. Учитывая отя‑ гощенный онкологический анамнез, на основании литературных данных о природе наследственно обусловленной УМ больной было выполнено ДНК‑тестирование выбранных для анализа генов‑ кандидатов – BRCA1 , BRCA2 и CHEK2 . Выяв‑ ленная патогенная мутация в гене CHEK2 ранее описана как возможная причина СЛФ и ЛФПС [30, 31]. В одном из исследований с целью оцен‑ ки вклада герминальных мутаций в гене CHEK2 в патогенез СЛФ Bell et al. представили четыре клинических наблюдения классического варианта данного синдрома [32]. Предварительный анализ показал отсутствие герминальных мутаций в гене ТР53 у всех пациентов. Наследуемые мутации в гене CHEK2 были выявлены у всех больных , миссенс‑вариант в 157 кодоне (подтвержденный в представленном нами клиническом наблюдении) был диагностирован в одном случае у пробанда с ПМЗН: колоректальным раком и билатеральной УМ, – что позволило авторам отнести герминаль‑ ные мутации в гене CHEK2 к возможным причинам развития СЛФ у пациентов.
Современные критерии для постановки клас‑ сического диагноза СЛФ и ЛФПС, предложенные Birch в 1994 г. [33] и дополненные Eeles годом позже [34], а также критерии Chompret для ТР53 ‑ тестирования [35] не позволяют нам с учетом
Список литературы Наследственная увеальная меланома: обзор литературы и клинический случай
- Harbour J.W. The genetics of uveal melanoma: an emerging framework for targeted therapy. Pigment Cell Melanoma Res. 2012 Mar; 25 (2): 171-81. DOI: 10.1111/j.1755-148X.2012.00979.x
- Helgadottir H., Hoio V. The genetics of uveal melanoma: current insights. Appl Clin Genet. 2016 Sep 6; 9: 147-55. DOI: 10.2147/TACG.S69210
- Nichols E.E., Richmond A., Daniels B. Tumor Characteristics, Genetics, Management, and the Risk of Metastasis in Uveal Melanoma. Semin Ophthalmol. 2016; 31 (4): 304-9. DOI: 10.3109/08820538.2016.1154175
- Singh A.D., Rennie I.G., Seregard S., Giblin M., McKenzie J. Sunlight Exposure and Pathogenesis of Uveal Melanoma. Surv Ophthalmol.2004; 49 (4): 419-28.
- Decatur C.L., Ong E., Garg N., Anbunathan H., Bowcock A.M., Field M.G., Harbour J.W. Driver Mutations in Uveal Melanoma: Associations With Gene Expression Profile and Patient Outcomes. JAMA Ophthalmol. 2016 Jul 1; 134 (7): 728-33. DOI: 10.1001/jamaophthalmol.2016.0903
