Новые N- [2- (бензоилфенокси) этил] производные нуклеиновых оснований - синтез и анти-ВИЧ-1 активность in vitro
![Новые N- [2- (бензоилфенокси) этил] производные нуклеиновых оснований - синтез и анти-ВИЧ-1 активность in vitro](/file/picture/142149260/novye-n2benzoilfenoksi-jetil-proizvodnye-nukleinovyh-osnovanijsintez-i.png "Новые N- [2- (бензоилфенокси) этил] производные нуклеиновых оснований - синтез и анти-ВИЧ-1 активность in vitro")
Автор: Озеров А.А., Новиков М.С., Луганченко А.И., Хартман Т., Букхайт Р.У.
Журнал: Волгоградский научно-медицинский журнал @bulletin-volgmed
Рубрика: Фармакология токсикология
Статья в выпуске: 4 (36), 2012 года.
Бесплатный доступ
Алкилированием триметилсилилпроизводных пиримидиновых оснований изомерными 1-бром-2-(бензоилфенокси)этанами синтезированы новые производные бензофенона. Орто-производные урацила и тимина продемонстрировали высокую анти-ВИЧ-1 активность in vitro с величиной EC 50 0,030 и 0,009 μМ, соответственно. Мета- и пара-изомеры были полностью неактивны.
Анти-вич-1 активность, пиримидин, бензофенон
Короткий адрес: https://sciup.org/142149260
IDR: 142149260 | УДК: 615.3:
Novel n- [2- (benzoylphenoxy) ethyl] nucleic bases derivatives - synthesis and anti-HIV-1 activity in vitro
Текст статьи Новые N- [2- (бензоилфенокси) этил] производные нуклеиновых оснований - синтез и анти-ВИЧ-1 активность in vitro
Ненуклеозидные ингибиторы обратной транскриптазы (ОТ) ВИЧ-1 пиримидиновой природы представляют собой наиболее перспективный класс современных лекарственных средств комплексной терапии ВИЧ-1 инфекции [2]. Среди соединений, продемонстрировавших высокий уровень противовирусной активности, следует выделить производные бензофенона, содержащие этот двуядерный ароматический фрагмент на конце ациклической цепи в N1-замещенных урацилах. Ранее нами были синтезированы производные 1-[2-(2-бензоилфенокси)эти-л]урацила, содержащие разнообразные заместители в бензофеноновом фрагменте. Некоторые из них оказались эффективными ингибиторами ОТ ВИЧ-1 и подавляли репродукцию различных штаммов виру- са in vitro в наномолярных концентрациях [6]. Однако родоначальная структура 1-[2-(2-бензоилфенокси)-этил]урацила, не содержащая каких-либо заместителей во фрагментах урацила и бензофенона, нами ранее получена не была. Кроме того, до сих пор остается не до конца ясным характер влияния природы нуклеинового основания и типа замещения в бензофеноновом фрагменте (орто-, мета- или пара-) на противовирусные свойства соединений данного класса. Решению этих актуальных вопросов и посвящена настоящая работа.
ЦЕЛЬ РАБОТЫ
Синтез и исследование анти-ВИЧ-1 активности in vitro новых производных нуклеиновых основа- ний, содержащих фрагменты бензофенона, отличающихся характером их присоединения к ациклической цепи.
МЕТОДИКА ИССЛЕДОВАНИЯ
Спектры ЯМР 1Н и 13С регистрировали на спектрометре Bruker AMXIII-400 в растворе диметилсуль-фоксида-D6, внутренний стандарт — ТМС. Интерпретацию спектров осуществляли с помощью лицензионной программы ACD/HNMR Predictor Pro 3.0 фирмы Advanced Chemistry Development (Канада). ТСХ выполняли на пластинах Sorbfil, элюент — 2-пропанол, проявление в парах иода. Температуры плавления измерены в стеклянных капиллярах на приборе Mel-Temp 3.0 (Laboratory Devices Inc., США).
Исследование анти-ВИЧ-1 активности in vitro проводили в культуре CEM-SS-клеток, которые суспендировали в культуральной среде в количестве 105 клеток/мл и инфицировали ВИЧ-1 (штамм HTLV-IIIB) при мультипликации инфекции, равной 0,2. Немедленно после инфицирования вирусом вносили растворы, содержащие различные концентрации исследуемого вещества в ДМСО, и инкубировали в течение 4 сут. при температуре 37 оС. Число живых клеток устанавливали по окончании инкубации при помощи бромида 3-(4,5-диметилтиазол-2-ил)-2,5-дифенилтетразо-лия, при этом определяли концентрацию вещества, которая на 50% защищала CEM-SS-клетки от цито-патического эффекта ВИЧ-1 (ЕС50).
Цитотоксичность соединений изучали параллельно в неинфицированных культурах клеток, при этом определяли концентрацию вещества, которая на 50% уменьшала количество живых CEM-SS-кле-ток (CС50). Расчетным путем определяли индекс селективности, являющийся отношением цитотоксической концентрации к ингибиторной концентрации: SI = CС50/ ЕС50 [4].
РЕЗУЛЬТАТЫ ИССЛЕДОВАНИЯ И ИХ ОБСУЖДЕНИЕ
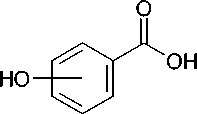
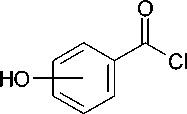
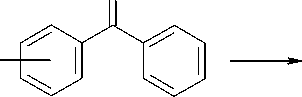
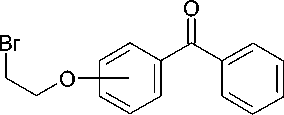
Синтез новых соединений был осуществлен, исходя из соответствующих изомеров оксибензой-ных кислот (салициловой, мета- и пара-оксибензой-ной), которые реакцией с хлористым тионилом превращали сначала в хлорангидриды, а затем по реакции Фриделя-Крафтса в орто-, мета- и пара-оксибензофеноны. Кипячение последних с 1,2-дибромэтаном в безводном ацетоне в присутствии карбоната калия привело к 2-бромэтиловым эфирам, которыми по разработанному нами ранее методу [1] алкилировали триметилсилилпроизводные пиримидиновых оснований — урацила, тимина и цитозина, а также калиевую соль аденина, получаемую in situ из аденина-основания и карбоната калия в среде безводного диметилформамида по известной методике (рис.) [3].
Выход и физико-химические свойства полученных соединений представлены в табл. 1.
SOCl2
C 6 H 6
AlCl3
O
BrCH2CH2Br
K2CO3
Me3Si-Base
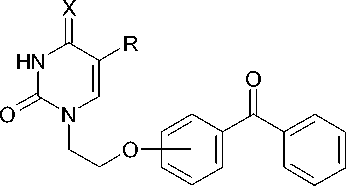
I - V
Adenine K2CO3
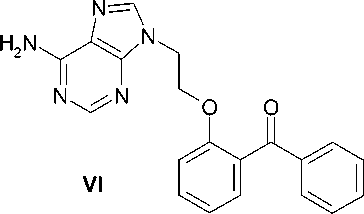
Рис. Синтез N-[2-(бензоилфенокси)этил]производных нуклеиновых оснований
ТАБЛИЦА 1
Свойства синтезированных соединений
|
Соединение |
X |
R |
Тип замещения |
Выход, % |
Брутто-формула |
Т. пл., оС |
R f |
|
I |
O |
H |
орто |
57 |
C 19 H 16 N 2 O 4 |
157,0 — 159,5 |
0,67 |
|
II |
O |
H |
мета |
59 |
C 19 H 16 N 2 O 4 |
209,5 — 212,0 |
0,60 |
|
III |
O |
H |
пара |
65 |
C 19 H 16 N 2 O 4 |
199,0 — 201,5 |
0,52 |
|
IV |
O |
CH 3 |
орто |
52 |
C 20 H 18 N 2 O 4 |
178 — 180 |
0,71 |
|
V |
NH |
H |
орто |
46 |
C 19 H 17 N 3 O 3 |
214 — 217 |
0,30 |
|
VI |
— |
— |
орто |
65 |
C 20 H 17 N 5 O 2 |
199 — 201 |
0,25 |
Исследование противовирусных свойств синтезированных соединений в отношении дикого штамма ВИЧ-1 показало, что родоначальное соединение — 1-[2-(2-бензоилфенокси)этил]урацил ( I ), не имеющее никаких заместителей в ароматических ядрах, тем не менее обладает высокой противовирусной активностью и эффективно защищает Т-лимфоциты от цитопатическо-го действия ВИЧ-1. Величина ингибиторной концентрации этого вещества составляет EC50 = 0,030 µM при отсутствии цитотоксических свойств во всем изученном диапазоне концентраций (табл. 2). По своему противовирусному действию это вещество более чем в 2 раза превосходит невирапин (EC50 = 0,075 µM [6]) и мало уступает самым активным из синтезированных нами ранее замещенных аналогов [6]. Изменение характера замещения во фрагменте бензофенона с орто- на мета-или пара- приводит к полному исчезновению противовирусных свойств и появлению заметной цитотоксичности (соединения II и III ). Таким образом, наличие ортотипа замещения во фрагменте бензофенона является наиболее критичным фактором, определяющим саму способность вещества подавлять репродукцию ВИЧ-1.
ТАБЛИЦА 2
Противовирусная активность синтезированных соединений
|
Соединение |
Ингибиторная концентрация EC 50 , µM |
Цитотоксическая концентрация СC 50 , µM |
Индекс селективности EC 50 / EC 50 |
|
I |
0,030 |
> 100 |
> 3333 |
|
II |
> 100 |
73,1 |
< 1 |
|
III |
> 100 |
79,2 |
< 1 |
|
IV |
0,009 |
> 100 |
> 11111 |
|
V |
> 100 |
> 100 |
< 1 |
|
VI |
0,670 |
> 100 |
> 149 |
Замена нуклеинового основания в 1-[2-(2-бензо-илфенокси)этил]урациле ( I ) на цитозин (соединение V ) или аденин (соединение VI ) приводит к исчезновению или значительному ослаблению противовирусной активности. В противоположность этому, тиминовый аналог (соединение IV ) продемонстрировал еще более выраженные противовирусные свойства с величиной EC50 = 0,009 µM. Это дает основание полагать, что введение и других заместителей в положение С5 пиримидиновой системы базовой молекулы 1-[2-(2-бен-зоилфенокси)этил]урацила ( I ) может привести к получению высокоактивных соединений.
ЗАКЛЮЧЕНИЕ
Таким образом, нами установлено, что в ряду пиримидиновых производных бензофенона наиболее важными факторами, определяющими наличие высокой анти-ВИЧ-1 активности, являются наличие ортозамещения во фрагменте бензофенона и карбонильного атома кислорода в положении С4 пиримидиновой системы (урацил или тимин). При этом введение дополнительного заместителя в положение С5 урацила приводит к многократному усилению противовирусных свойств. Результаты исследований, в сочетании с полученными нами ранее данными о характере влияния различных заместителей в ароматических ядрах бензофенона на уровень эффективной концентрации in vitro и способность веществ данного ряда ингибировать ОТ ВИЧ-1, могут быть использованы для дальнейшего направленного синтеза перспективных анти-ВИЧ-1 агентов пиримидиновой природы.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
1-[2-(2-Бензоилфенокси)этил]урацил (I).
-
( А ) Хлорангидрид салициловой кислоты. К 20,0 мл (0,275 моль) тионилхлорида добавляют 1,0 мл (0,013 моль) диметилформамида и при интенсивном перемешивании при температуре 5—10 оС порциями в течение 1 ч вносят 25,0 г (0,156 моль) тонкоизмельченного безводного салицилата натрия. Реакционную массу выдерживают в течение 1 сут. при комнатной температуре, избыток тионилхлорида отгоняют в вакууме водоструйного насоса при температуре бани не выше 60 оС, к остатку добавляют 200 мл н-гексана, фильтруют, фильтрат упаривают в вакууме при температуре не выше 60 оС и получают 19,5 г хлорангидрида салициловой кислоты в виде светло-желтой подвижной жидкости, выход 80 %.
( Б ) 2-Оксибензофенон. В трехгорлый реактор, снабженный обратным холодильником, термометром, капельной воронкой и эффективной мешалкой, помещают 100 мл (1,125 моль) бензола, 20,0 г (0,150 моль) безводного хлорида алюминия и при интенсивном перемешивании при температуре не выше 25 оС добавляют в течение 30 мин раствор 10,0 г (0,064 моль) хлорангидрида салициловой кислоты в 25 мл бензола. Реакционную массу перемешивают при температуре 55—60 оС в течение 2 ч, охлаждают до комнатной температуры и выливают при перемешивании в смесь 250 г воды со льдом. Добавляют
100 мл хлороформа, органический слой отделяют, промывают 5%-м раствором карбоната натрия, водой, сушат сульфатом магния, фильтруют и упаривают на кипящей водяной бане в вакууме водоструйного насоса. Остаток кристаллизуют из смеси н-гексан — диэтиловый эфир (9:1) и получают 9,9 г 2-оксибензофено-на, Т. пл. 38—39 оС (Т. пл. 38—40 оС [5]), выход 78 %.
-
( В ) 1-Бром-2-(2-бензоилфенокси)этан. Смесь 7,5 г (0,038 моль) 2-оксибензофенона, 15,0 мл (0,174 моль) 1,2-дибромэтана, 7,7 г (0,054 моль) безводного карбоната калия, 0,5 г (0,001 моль) дибензо-18-краун-6 и 150 мл безводного ацетона кипятят в течение 1 сут., фильтруют и упаривают на кипящей водяной бане в вакууме водоструйного насоса. Остаток растворяют в 100 мл хлороформа, промывают 5%-м раствором едкого натра, водой, 5%-м раствором хлористоводородной кислоты, водой, сушат сульфатом магния, фильтруют и упаривают на кипящей водяной бане в вакууме водоструйного насоса. Получают 9,8 г 1-бром-2-(2-бензоилфенокси)этана в виде светло-желтой вязкой жидкости, выход 85 %.
Спектр ЯМР 1Н, δ, м.д.: 3,83 т (2 Н, 4 Гц, СН2-Br); 4,42 т (2 Н, 4 Гц, СН2-N); 7,06-7,70 м (9 Н, арил).
( Г ) 1-[2-(2-Бензоилфенокси)этил]урацил. 2,0 г (0,018 моль) урацила, 25 мл (0,120 моль) гексаметилдисилазана и 0,25 г (0,005 моль) хлорида аммония кипятят с защитой от влаги воздуха до полного растворения осадка (4 ч), избыток гексаметилдисилазана отгоняют на кипящей водяной бане при остаточном давлении не менее 10 мм рт. ст., к остатку добавляют 3,0 г (0,010 моль) 1-бром-2-(2-бензоилфенокси)этана и нагревают при периодическом перемешивании при температуре 175—180 оС в течение 4 ч. Реакционную массу охлаждают, растворяют в 25 мл этилацетата, добавляют 25 мл 2-пропанола, через 30 мин выделившийся осадок отфильтровывают и фильтрат упаривают в вакууме. Остаток растворяют в 50 мл хлороформа, фильтруют, фильтрат упаривают в вакууме, остаток кристаллизуют из 50 мл 2-пропанола и получают 1,9 г 1-[2-(2-бензоилфенокси)этил]урацила, Т. пл. 157— 159,5 оС, выход 57%.
Спектр ЯМР 1Н, δ, м.д.: 3,76 т (2 Н, 5 Гц, СН2-N); 4,15 т (2 Н, 5 Гц, СН2-O); 5,18 д (1 Н, 8 Гц, Н5); 6,74 д (1 Н, 8 Гц, Н6); 7,06-7,71 м (9 Н, арил); 11,13 с (1 Н, NH)
Спектр ЯМР 13С, δ, м.д.: 47,09; 65,80; 100,48; 112,83; 121,19; 128,59; 129,21; 132,12; 133,45; 137,09; 145,39; 150,73; 155,73; 163,53; 195,53.
Соединения II - VI получают аналогично.
1-[2-(3-Бензоилфенокси)этил]урацил (II).
Спектр ЯМР 1Н, δ, м.д.: 4,08 т (2 Н, 5 Гц, СН2-О); 4,25 т (2 Н, 5 Гц, СН2-N); 5,56 д (1 Н, 6,5 Гц, Н5); 7,25-7,72 м (10 Н, арил, Н6 ); 11,32 с (1 Н, NH).
Спектр ЯМР 13С, δ, м.д.: 50,28; 69,00; 104,07; 118,41; 122,29; 125,94; 131,92; 132,96; 136,17; 140,31; 149,63; 154,36; 161,36; 167,11; 198,81.
1-[2-(4-Бензоилфенокси)этил]урацил (III).
Спектр ЯМР 1Н, δ, м.д.: 4,02 т (2 Н, 5 Гц, CH2-N); 4,21 т (2 Н, 5 Гц, СН2-О); 5,5 дд (1 Н, 8 Гц, 8 Гц, Н5); 6,99 -7,62 м (10 Н, арил, Н6); 11,28 с (1 Н, NH).
Спектр ЯМР 13С, δ, м.д.: 42,30; 63,70; 97,60; 105,12; 114,40; 128,70; 129,78; 129,60; 132,50; 137,80; 151,40; 153,80; 160,40; 162,60; 196,10.
1-[2-(2-Бензоилфенокси)этил]тимин (IV).
Спектр ЯМР 1Н, δ, м.д.: 1,57 с (3 Н, СН3): 3,75 т (2 Н, 5 Гц, СН2-N); 4,15 т (2 Н, 5 Гц, СН2-O); 6,79 с (1 Н, Н6); 7,06-7,69 м (9 Н, арил); 11,20 с (1 Н, NH).
Спектр ЯМР 13С, ?, м.д.: 11,90; 46,84; 65,84; 107,98; 112,91; 121,10; 128,55; 129,17; 132,09; 133,40; 141,50; 150,72; 155,45; 164,13; 195,36.
1-[2-(2-Бензоилфенокси)этил]цитозин (V).
Спектр ЯМР 1Н, δ, м.д.: 3,72 т (2 Н, 7 Гц, СН2-N); 4,07 т (2 Н, 7 Гц, СН2-O); 7,09-7,76 м (13 Н, арил, Н5, Н6, NH2).
Спектр ЯМР 13С, δ, м.д.: 48,20; 65,30; 93,19; 113,02; 121,31; 132,07; 133,55; 136,84; 146,49; 148,40; 149,67; 155,22; 161,28; 195,47.
9-[2-(2-Бензоилфенокси)этил]аденин (VI).
Спектр ЯМР 1Н, δ, м.д.: 4,15 т (2 Н, 5 Гц, CH2-N); 4,25 т (2 Н, 5 Гц, CH2-O); 7,01-7,56 м (11 Н, арил, Н3,Н8); 8,02 с (2 Н, NH2).
Спектр ЯМР 13С, δ, м.д.: 40,20; 65,50; 112,30; 125,22; 127,4; 128,07; 130,30; 132,38; 133,47; 138,78; 139,20; 140,17; 150,30; 156,23; 157,67; 192,34.
