Об использовании синхронного термоанализатора Netzsch 449 Jupiter для определения точки кипения веществ

Автор: Жеребцов Дмитрий Анатольевич, Толстогузов Дмитрий Сергеевич, Буланова Александра Владимировна, Авдин Вячеслав Викторович
Журнал: Вестник Южно-Уральского государственного университета. Серия: Химия @vestnik-susu-chemistry
Рубрика: Физическая химия
Статья в выпуске: 2 т.15, 2023 года.
Бесплатный доступ
Исследовано влияние массы воды, п- ксилола, этиленгликоля, нафталина на измеряемое значение температур и теплот их превращений. Показано, что синхронный термоанализатор Netzsch 449F1 Jupiter можно успешно использовать для определения точки кипения веществ. Температуры окончания их испарения монотонно возрастают с ростом массы вещества. Показано, что с достижением определенной массы процесс кипения становится стационарным, сопровождающимся формированием линейного фронта пика кипения, близкого по наклону к наклону фронта пиков плавления различных чистых веществ. Предложена и апробирована модифицированная методика обработки данных дифференциальной сканирующей калориметрии для решения этой задачи, дающая погрешность определения точки кипения ±4 °С в случае воды, этиленгликоля и п-ксилола и ±6 °С в случае нафталина. Погрешность определения теплот кипения по данным синхронного термоанализатора составляет ±5 % в случае воды и нафталина, однако в случае этиленгликоля и п- ксилола погрешность достигает 10 и 40 % соответственно, что делает данный метод не количественным, а лишь полуколичественным или качественным в отношении определения теплот превращений. На примере антраниловой кислоты и дииодо- пара -ксилола проведена апробация метода и получены данные о температурах и теплотах превращений для данных соединений, причем теплоты превращений ранее не были известны в литературе. Так, антраниловая кислота по нашим данным имеет не описанное ранее полиморфное превращение при 91 °С с теплотой 28,5 Дж/г, плавление при 145,5 °С с теплотой 150 Дж/г и кипение при 230 °С с теплотой 310 Дж/г. Дииод- пара -ксилол по нашим данным плавится при 102 °С с теплотой 60 Дж/г и кипит при 310 °С с теплотой 95 Дж/г.
Термический анализ, точка кипения, точка плавления
Короткий адрес: https://sciup.org/147240923
IDR: 147240923 | УДК: 544-971+544.03+544.08+54.084 | DOI: 10.14529/chem230213
On using a Netzsch 449 Jupiter synchronous thermal analyzer to determine the boiling point of substances
The influence of the mass of water, p -xylene, ethylene glycol, and naphthalene on the measured values of temperature and heat of their transformations has been studied. It has been shown that a synchronous thermal analyzer Netzsch 449F1 Jupiter can be successfully used to determine boiling points of substances. The final temperatures of their evaporation monotonously increase together with an increase in the mass of a substance. It has been shown that with the achievement of a certain mass, the boiling process becomes stationary, accompanied by the formation of a linear front of the boiling peak, close in slope to the slope of the front of the melting peaks of various pure substances. A modified technique of differential scanning calorimetry data processing has been proposed and tested to solve this problem, giving an uncertainty in determining the boiling point of ± 4 °C in the case of water, ethylene glycol, and p -xylene, as well as ±6 °С in the case of naphthalene. The error in determining the boiling heats by the data of a synchronous thermal analyzer is ±5 % in the case of water and naphthalene; however, in the case of ethylene glycol and p-xylene, the error reaches 10 % and 40 %, respectively, which makes this method not quantitative, but only semi-quantitative or qualitative in regard to determination of the transformation heat. Using the example of anthranilic acid and diiodoparaxylene, the method has been tested, and the obtained data on temperatures and heats of transformations for these compounds; notably, the transformation heats were not previously known from the literature. For example, anthranilic acid, according to our data, has a previously undescribed polymorphic transformation at 91 °C with the heat of 28.5 J/g, melting at 145.5 °C with the heat of 150 J/g, and boiling at 230 °C with the heat of 310 J/g. According to our data, diiodoparaxylene melts at 102 °C with the heat of 60 J/g and boils at 310 °C with the heat of 95 J/g.
Текст научной статьи Об использовании синхронного термоанализатора Netzsch 449 Jupiter для определения точки кипения веществ
Синхронный термический анализ широко используется для определения температур твердофазных превращений веществ (превращения I и II рода, термолиз), а также температур их плавления. Однако применение данного метода для измерения точек кипения веществ до сих пор не является общепринятым и вызывает споры [1–5].
В работах [6, 7] авторами изучалась возможность применения методов ДСК и ТГА для определения температур плавления и кипения веществ. Авторы указали на особенности проведения эксперимента и показали факторы, изменяющие погрешность измерения. Обе работы подтвердили возможность измерения температур плавления и кипения с помощью оборудования термического анализа. Авторы [8–10] изучали возможность применения ТГ, ДСК и ДТГ для определения точек плавления, кипения и давления паров различных органических веществ. По результатам работ авторы сошлись во мнении, что данным методом можно определять указанные параметры, несмотря на незначительные отклонения в значении температур от литературных данных. В работах [11–15] авторы измеряли точки плавления и кипения органических и неорганических веществ методами ДСК и ДТА. Полученные результаты мало отличались от литературных данных.
Данная работа посвящена исследованию возможности использования синхронного термоанализатора Netzsch 449F1 Jupiter для определения точки кипения вещества.
Методика измерений
Все исследования были проведены в наиболее часто применяемом в работе термоанализатора режиме: скорость нагревания 10 °С/мин, атмосфера – аргон (суммарный расход 60 мл/мин), материал тигля – корунд. Перед проведением нагревания с указанной скоростью образцы нагрева- лись до 30 °С и выдерживались при этой температуре 10 мин, что улучшало воспроизводимость стартовых условий эксперимента.
В корундовый тигель дозировалась дистиллированная вода, после чего тигель накрывался крышкой с микроотверстием и помещался в измерительную ячейку. Ввиду испарения небольшой части воды в процессе пробоподготовки, загрузки и выдержки при 30 С в дальнейших расчетах применялась не масса навески, определенная на весах перед загрузкой, а масса, определенная по кривой термогравиметрии (ТГ), что исключало погрешность, связанную с испарением воды до начала нагревания. В процессе обработки полученных кривых ТГ и дифференциальной сканирующей калориметрии (ДСК) из исходных кривых вычитались базовые кривые, полученные в тех же условиях с пустым тиглем образца. Этот стандартный прием позволял исключить эффекты, связанные с конвекцией газа в печи и нестационарными тепловыми процессами при старте нагревания.
На рис. 1 представлена серия кривых ТГ, на рис. 2 – серия кривых ДТГ, а на рис. 3 – серия кривых ДСК, полученные с разными по массе образцами воды, от 2,4 до 26 мг. Для определения точки окончания испарения воды использовались два метода: с помощью кривых ДТГ или ДСК. В обоих случаях выбиралось положение наиболее высокотемпературного минимума на кривой, что соответствовало испарению последних микрограмм воды.
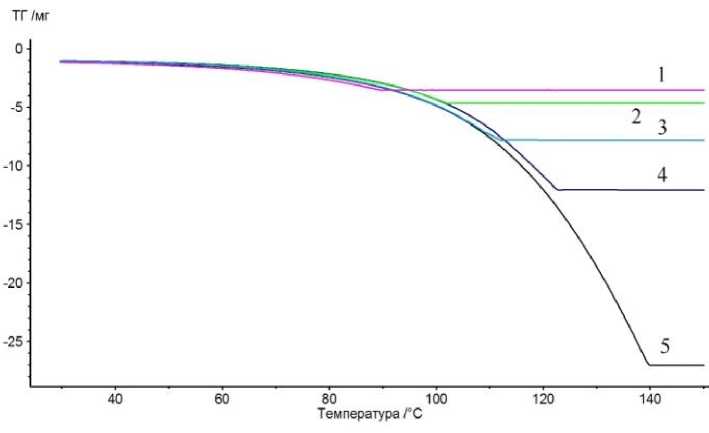
Рис. 1. Кривые ТГ навесок воды разной массы: 1 – 2,4 мг, 2 – 3,6 мг, 3 – 6,7 мг, 4 – 11 мг, 5 – 26 мг
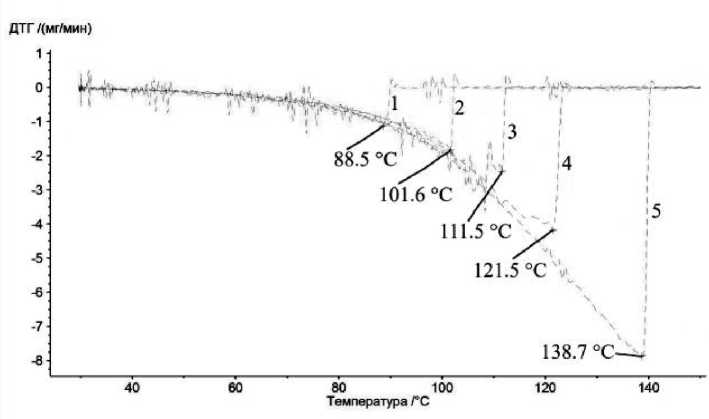
Рис. 2. Кривые ДТГ навесок воды разной массы: 1 – 2,4 мг, 2 – 3,6 мг, 3 – 6,7 мг, 4 – 11 мг, 5 – 26 мг
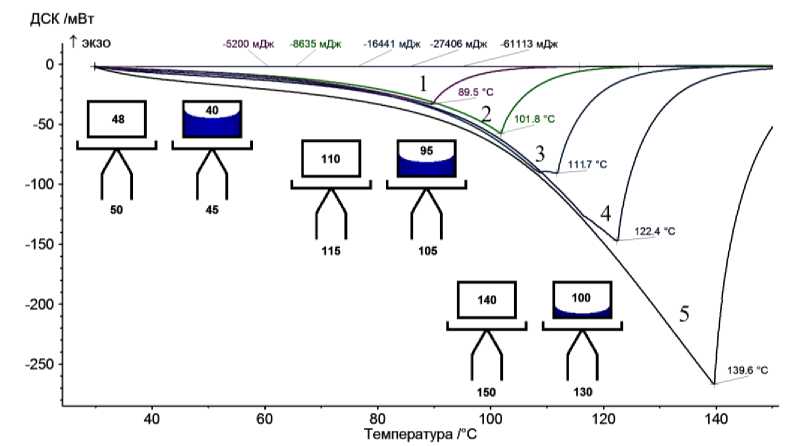
Рис. 3. Кривые ДСК навесок воды разной массы. На врезках – схемы распределения температур в ходе нагревания измерительной ячейки: слева – тигель сравнения, справа – тигель с водой. Числа на схемах соответствуют градусам Цельсия. Температура нагревателя на схемах слева направо: 60, 125 и 160 °С
Наблюдаемые температуры окончания испарения приведены в зависимости от массы испарившейся воды на рис. 4. Два набора точек соответствуют температурам, определенным по минимумам на кривых ДТГ или ДСК.
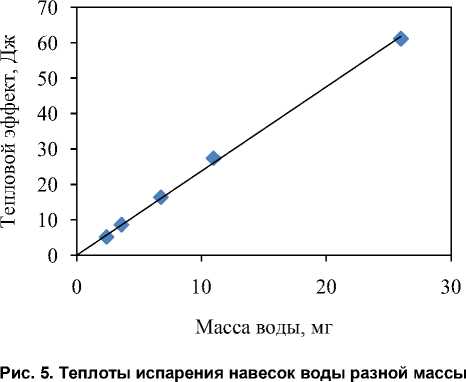
Определенные по площади пиков ДСК теплоты превращений очень хорошо коррелируют с массой испарившейся воды (рис. 5). Вычисленная из этого графика теплота испарения воды 2375 Дж/г (в диапазоне от 30 до 100 °С) хорошо согласуется со справочным значением 2430 Дж/г (при 30 °С) и 2258 Дж/г (при 100 °С) [16].
♦ ДТ Г
0 10 20 30
Масса воды, мг
Рис. 4. Температуры окончания испарения навесок воды разной массы, определенные по пикам ДТГ и ДСК. Точка при 19 мг соответствует эксперименту в платиновом тигле
Из анализа данных (см. рис. 1–4) следует, что измеренная температура окончания испарения может быть как выше, так и ниже температуры кипения жидкости. В случае воды в исследованном диапазоне масс этот диапазон составляет от 88 до 140 °С. Столь большой диапазон разительно отличается от точности определения температур плавления веществ, составляющей не более 0,5–1 градуса в данной области температур.
Для использования синхронного термоанализа в качестве инструмента для определения температуры кипения вещества необходимо ввести дополнительные ограничения к предлагаемому методу. Выяснить, какие же это ограничения, позволяет анализ тепловых процессов внутри измерительной ячейки. Рассмотрим эти процессы в динамике нагревания.
Внутри печи синхронного термоанализатора в процессе измерения в режиме линейного нагрева устанавливается определенное распределение температур. Наиболее горячим объектом термоанализатора является проволочный нагреватель печи. Его теплота передается более холодному корпусу трубчатой печи. Печь, в свою очередь, передает теплоту более холодным деталям измерительной ячейки: термопарному столику, термопаре в корундовой соломке, а также тиглю. И наконец, последним воспринимает теплоту от тигля образец, находящийся внутри него и являющийся наиболее холодным объектом в рассматриваемой системе (схемы на рис. 3).
При приближении к точке кипения внутри тигля (под крышкой с микроотверстием) состав атмосферы приближается к насыщенному пару жидкости (поток пара постепенно вымывает молекулы воздуха или аргона из пространства внутри тигля). Поток пара из тигля иллюстрируется термогравиметрическими кривыми, на которых задолго до конца испарения потеря массы воды составляет вне зависимости от массы образца одинаковую величину, например, 1,1–1,5 мг при 80 °С (см. рис. 1). Это обусловлено идентичностью состава атмосферы в тигле и вокруг него, а также воспроизводимыми условиями массообмена (одинаковый зазор между крышкой и стенками тигля, постоянный диаметр отверстия в крышке тигля). Данная геометрия тигля и ячейки приводит к закономерному уносу воды из всех образцов по одному закону (см. рис. 1). В случае слишком маленькой навески унос паров воды приводит к тому, что, не достигнув 100 °С, вся навеска успевает в виде пара перейти из тигля в печное пространство.
В случае больших навесок воды выступает на первое место фактор сравнительно низкой теплопроводности среды между термопарой и образцом. Как отмечено выше, теплота, поступающая от нагревателя внутрь печи, в первую очередь вызывает нагрев термопары как более теплопроводной, легкой и наименее теплоемкой части системы. От образца термопара отделена стенкой тигля и воздушным зазором между тиглем и термопарным столиком (столик имеет неровности и шероховатости, что обуславливает лишь точечный его контакт с дном тигля). В результате при задержке роста температуры образца внутри тигля или даже полной остановке этого роста (в случае протекания в образце нонвариантных превращений – плавления, кипения и т. п.) температура термопары, регистрируемая термоанализатором как температура образца, продолжает расти (рис. 3).
Тем не менее, в случае проведения эксперимента по определению точки кипения воды в платиновом тигле (точка при 19 мг на рис. 4) погрешность оказывается того же порядка. Это означает, что в общее тепловое сопротивление вклад от воздушного зазора между термопарным столиком и дном тигля намного больше, чем вклад, обусловленный теплопроводностью стенки тигля.
В случае навески воды массой более 3,5 мг наблюдается завышенное значение температуры окончания испарения (см. рис. 1–4). Несомненно, что при этом в процессе измерения температу-
Жеребцов Д.А., Толстогузов Д.С., Об использовании синхронного термоанализатора Буланова А.В., Авдин В.В. Netzsch 449 Jupiter… ра воды растет, достигая 100 °С и оставаясь на этом значении вплоть до полного ее испарения. Однако тепловой поток от нагревателя успевает перегреть термопару образца на десятки градусов (см. рис. 3). Столь большая величина обусловлена рядом причин.
Во-первых, это низкая скорость радиационного теплообмена при 100-150 °С. С ростом температуры вклад от передачи тепла излучением между стенкой тигля и термопарным столиком (через воздушный зазор) значительно уменьшает разницу между температурой образца и термопары. Измерение температур кипения при более высоких температурах приведет к меньшим погрешностям.
Во-вторых, особенностью воды является то, что она имеет в несколько раз более высокую теплоту кипения на единицу массы или единицу объема, чем другие жидкости. Измерение температур кипения других жидкостей потребует более массивных навесок.
Следует подчеркнуть, что причиной большой задержки завершения испарения является именно большая величина теплоты, которую необходимо передать внутрь тигля. Магическая масса навески воды 3,5 мг, измеренная температура окончания испарения которой практически совпадает с температурой кипения воды, отнюдь не случайна. Хорошо известно, что теплота плавления большинства веществ на порядок ниже теплоты испарения. Поэтому теплота, поглощаемая при испарении 3,5 мг воды близка по величине к теплоте, поглощаемой при плавлении, например, навески 50 мг бензойной кислоты, по которой проводится калибровка термоанализатора. Это обуславливает и примерно равную задержку во времени (и соответствующую разницу в измеренной температуре) для навески кипящей воды или плавящейся бензойной кислоты, если точку превращения этих веществ сравнивать по положению максимума пика ДСК.
Наиболее простым подходом к поиску точки кипения может служить обработка пика испарения по такому же алгоритму, что и поиск точки плавления чистого вещества. Действительно, при анализе группы кривых на рис. 3 можно для максимальной навески (26 мг воды) провести касательную к низкотемпературной ветви пика вблизи его минимума (через точку перегиба) и построить стандартным методом (применяемым для определения начала пика плавления) точку начала превращения (команда Onset в программном пакете Netzsch Proteus), когда за базовую линию выбирается касательная в точке до крутого снижения кривой ДСК (50-60 °С). Это приводит к пересечению двух этих касательных при температурах, близких к точке кипения воды (рис. 6): 103,6 °С (26 мг), 97,9 °С (11 мг).
Важным ограничением для такого подхода является применение заведомо большой массы навески, многократно превышающей массу, испаряющуюся без перегрева термопары выше точки кипения. При проведении аналогичной операции с меньшими навесками будут получаться неоправданно заниженные значения: 92,7 °С (6,7 мг), 91,3 °С (3,6 мг), 74,9 °С (2,4 мг).
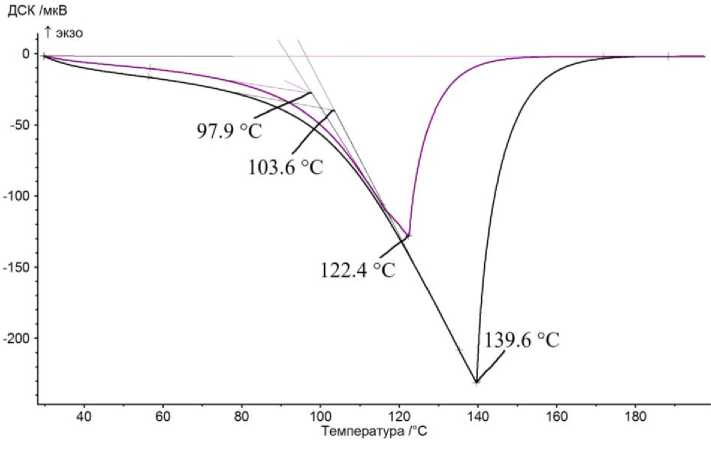
Рис. 6. Обработка кривых ДСК стандартным способом построения точки начала превращения (Onset)
Причину такого поведения кривых ДСК легко понять, сравнивая пики испарения воды с пиками плавления чистого вещества, например, бензойной кислоты, индия и бифенила С12Н10 (рис. 7). Оказывается, что для них величины крутизны фронта пиков плавления близки между собой. В точке перегиба они составляют соответственно –56,1 и –54,5 и –52,2 мкВ/мин по данным дифференцирования кривой ДСК (массы навесок составляли 15,3, 20,5 и 14,4 мг). Для воды эти значения несколько меньше и быстро убывают с уменьшением массы навесок: –46,4 (26 мг), –41,7 (11 мг), –32,1 (6,7 мг), –28,0 (3,6 мг) и –9,7 мкВ/мин (2,4 мг). Связано это с тем, что при большой навеске воды (например, 26 мг) процесс ее испарения в большей мере протекает в изотермическом режиме при температуре 100 °С. То есть в том же режиме фазового превращения I рода, что и плавление чистых веществ. В результате фронт пика испарения воды массой 26 мг имеет длинный прямой участок, как и пик плавления, например, бензойной кислоты. Действительно, масса воды, испаряющейся к моменту достижения термопарой температуры 100 °С, составляет около 3,5 мг. Масса воды, превышающая эту величину, ведет себя в режиме беспузырькового кипения в точности, как плавящаяся бензойная кислота, сохраняя температуру образца постоянной.
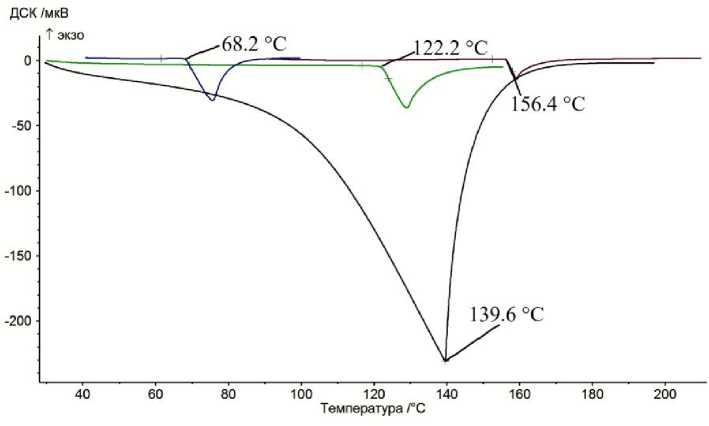
Рис. 7. Кривые ДСК бифенила С 12 Н 10 , воды, бензойной кислоты и индия
Обобщая сделанные выводы, можно предложить метод для определения температуры кипения веществ. Для этого сначала проводится эксперимент с нагреванием исследуемого вещества до температуры, превышающей температуру окончания его испарения на 50–70 °С (до возвращения кривой ДСК к исходной базовой линии). В случае, если на фронте пика испарения вещества имеется линейный участок, составляющий 40–50 % от высоты пика, проводится построение точки кипения стандартным методом поиска точки начала плавления. При этом точку, через которую следует провести касательную к базовой линии, следует выбрать на участке, отстоящем на 3–4 температурных интервала ниже, чем интервал линейного участка.
Например, на кривой испарения навески воды массой 26 мг линейный участок занимает приблизительно интервал 120–140 °С, а точка, через которую проведена касательная к базовой линии, выбрана при 55 °С (см. рис. 6). Одновременно определяется значение производной от кривой ДСК в точке перегиба. Это значение должно быть близко (в пределах 20 %) к значению производной от кривой ДСК в точке перегиба пика плавления чистого вещества в близком интервале температур (±50 °С). В случае, если линейный участок слишком короток, а также производная от кривой ДСК в точке перегиба пика испарения намного меньше, чем для пика плавления, следует перейти к построению касательной к ветви фронта другим способом. Для этого в точке минимума на пике ДСК следует построить линию, имеющую то же значение производной от кривой ДСК, что и пик плавления чистого вещества. В случае навески воды 6,7 мг это приводит к значению температуры кипения 99–102 °С. При меньших навесках этот метод также способен дать приемлемую точность: 96,6 °С (3,6 мг) (рис. 8). И лишь при навесках воды, испаряющихся полностью до достижения температуры кипения, этот метод непригоден: 87 °С (2,4 мг).
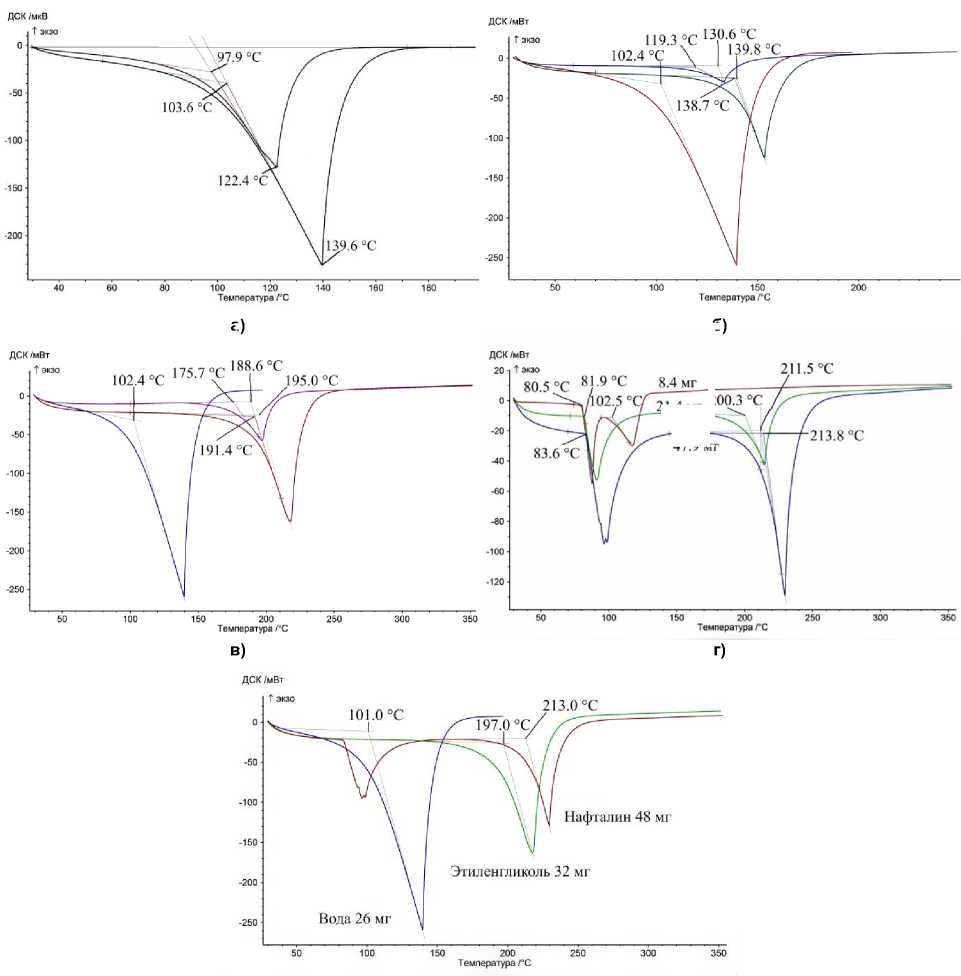
б)
а)
47.9 мг
д)
Рис. 8. Применение предложенного метода определения температуры кипения на примере навесок воды 26 и 3,6 мг (а), п- ксилола 12,5 и 42,2 (б), этиленгликоля 10,5 и 31,5 (в), нафталина 8,4, 21,4 и 47,9 мг (г), объединенный график на максимальных навесках (д). На графики ксилола и этиленгликоля добавлены кривые ДСК воды для сравнения
21.4 мг
Корректность метода определения температуры кипения может быть продемонстрирована на примере воды, ксилола, этиленгликоля и нафталина (табл. 1). При работе в одинаковых условиях (материал тигля, скорость нагрева, состав атмосферы и скорость потока газа) можно выявить зависимость крутизны фронта пика плавления чистых веществ от температуры (производной кривой ДСК или ДДСК) и в дальнейшем пользоваться усредненным интерполированным значением этой производной (рис. 9). На рисунке приведены значения производных для 27 веществ (табл. 2), по два значения приведены для навесок разной массы. Меньшие значения производных фронта плавления (30–40 мкВ/мин для 30–500 °С) чаще всего связаны с более пологим фронтом пика, вызванным более низкой чистотой или высокой пористостью (низкой теплопроводностью) образца. Немногочисленные примеры более высоких производных (60–70 мкВ/мин для 30– 500 °С) могут быть связаны с протекающими параллельно с плавлением реакциями термолиза, например, декарбоксилирования карбоновых кислот или потери воды фталевой кислотой.
Таблица 1
Температуры и теплоты превращений воды, ксилола, этиленгликоля, нафталина, антраниловой кислоты и дииод- пара -ксилола на навеске 20–50 мг
|
Вещество |
Температура, °С |
Теплота, Дж/г |
Источник |
|
Вода |
кип. 97–104 кип. 100 |
кип. 2200–2400 (среднее на 30–100 °С) кип. 2430 и 2258 (30 и 100 °С) |
[16] |
|
п- Ксилол |
кип. 138–140 кип. 138,4 |
кип. 240–250 кип. 400 (42,40 кДж/моль) |
[17] |
|
Этиленгликоль |
кип. 189–197 кип. 197,6 |
кип. 715–865 кип. 683 (58,71 Дж/моль) |
[18] |
|
Нафталин |
пл. 80,5–83,6 пл. 80,3 кип. 212–214 кип. 218 |
пл. 175–185 пл. 150,7 (19,288 кДж/моль) кип. 330–340 кип. 339,8 (43,5 кДж/моль) |
[19] |
|
Антраниловая кислота |
полим. 90–92 пл. 145–146 пл. 145–147 кип. 220–250 кип. – |
полим. 27–30 пл. 130–170 пл. – кип. 290–330 кип. – |
[20, 21] |
|
Дииод- пара -ксилол |
пл. 100–103 пл. 103–104 кип. 280–340 |
пл. 55–65 пл. – кип. 90–100 |
[22] |
Таблица 2
Производные фронта плавления или кипения, мкВ/мин
|
Вещество |
Т, °С |
ДДСК, мкВ/мин |
Масса, мг |
|
Бифенил |
69 |
57,9 |
11,2 |
|
8-оксихинолин |
73 |
41,8 |
9,4 |
|
Нафталин |
80 |
54,1 |
8,8 |
|
Нафталин |
80 |
49,1 |
47,2 |
|
Фенилдиацетилентриметилсилан |
86 |
49,2 |
7,9 |
|
Вода |
100 |
44,8 |
19,2 |
|
Вода |
100 |
46,4 |
26,4 |
|
Дииод- пара -ксилол |
103 |
45,2 |
17,1 |
|
Дипропаргиловый спирт |
108 |
35,3 |
4,9 |
|
Бензойная к-та |
122 |
48,5 |
6,3 |
|
Бензойная к-та |
122 |
56,1 |
13,9 |
|
п- Ксилол |
139 |
56,3 |
42,5 |
|
Антраниловая к-та |
145 |
63,8 |
41,4 |
|
Антраниловая к-та |
145 |
51 |
14,6 |
|
In |
156 |
58,3 |
7,0 |
|
In |
156 |
38,2 |
6,9 |
|
Салициловая к-та |
157 |
47,5 |
7,6 |
|
RbNO 3 |
163 |
32,2 |
5,2 |
|
п -Аминобензойная к-та |
188 |
56,4 |
3,4 |
|
Этиленгликоль |
197 |
46,2 |
32,9 |
|
Фталевая к-та |
215 |
67 |
14,4 |
|
Sn |
232 |
28,6 |
35,2 |
|
Бис (метилсалицил)диацетилен |
246 |
56,6 |
7,9 |
|
Тетраиодпараксилол |
254 |
43,4 |
14,1 |
|
Бис (бромфенил)диацетилен |
269 |
60,5 |
6,7 |
|
KClO 4 |
300 |
52,2 |
10,6 |
|
Bi |
272 |
64,5 |
35,2 |
|
2,3-7,8-дибензпирен-1,6-хинон |
364 |
48,6 |
7,5 |
Окончание табл. 2
|
Вещество |
Т, °С |
ДДСК, мкВ/мин |
Масса, мг |
|
Zn |
419 |
60,3 |
9,4 |
|
цис -Перинон |
471 |
40,5 |
7,7 |
|
CsCl |
472 |
23,4 |
11,2 |
|
Al |
660 |
21,1 |
4,1 |
|
K 2 CrO 4 |
669 |
23,2 |
3,1 |
|
BaCO 3 |
809 |
47,2 |
16,3 |
|
BaCO 3 |
809 |
30,7 |
4,4 |
|
Ag |
950 |
34 |
14,7 |
|
Au |
1062 |
33,9 |
22,8 |
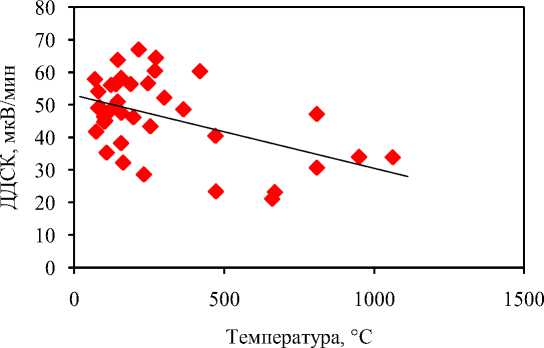
Рис. 9. Зависимость значения производной кривой ДСК в точке перегиба ветви фронта пика плавления чистых веществ, измеренных в аргоне в корундовых тиглях при скорости нагревания 10 °С/мин, от температуры превращения
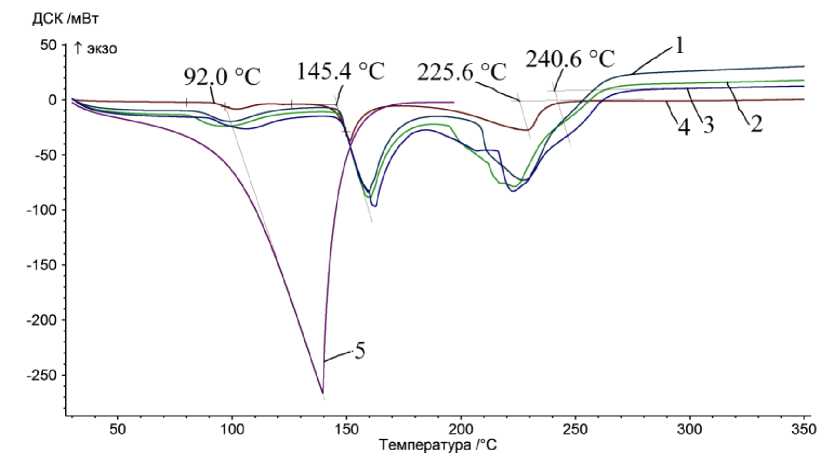
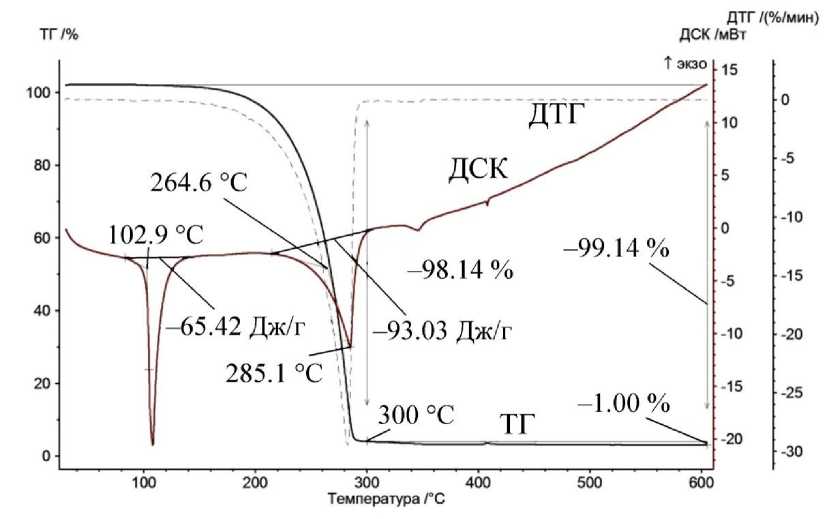
Апробация метода может быть проведена на примере антраниловой кислоты и дииодо- пара -ксилола (рис. 10). Литературные данные по антраниловой кислоте противоречивы, указывают на отсутствие точки кипения (наличие возгонки) и протекание процесса возгонки [21, 23]. По источнику [23] при нагревании в интервале температур 197 ± 3 °С при скорости нагрева 10 К/мин происходит декарбоксилирование антраниловой кислоты. При этом потеря массы составляет 94,5 %. Согласно [24], антраниловая кислота имеет три полиморфные структуры и способна к образованию димеров. Согласно полученным данным, антраниловая кислота при 92 °С испытывает полиморфное превращение (теплота превращения 29 Дж/г), при 145,5 °С плавится (теплота плавления 128 Дж/г) и кипит около 240 °С (теплота испарения 302 Дж/г). Дииодо- пара -ксилол при 102,9 °С плавится (теплота плавления 65 Дж/г) и кипит около 265 °С (теплота испарения 93 Дж/г). По литературным данным, дииодо- пара -ксилол плавится при 103–104 °С [23]. На примере нафталина (рис. 8г) можно сделать вывод, что для легколетучих веществ следует особенно следить за тем, чтобы наклоны фронта пиков плавления и кипения были близки, чтобы определение температуры кипения было корректным. Антраниловая кислота при увеличении навески проявляет невоспроизводимые отклонения от ожидаемой формы пика кипения, что может говорить о протекании параллельно с испарением процессов разложения, вспенивания или полимеризации. Таким образом, достоверные результаты можно получить лишь для процессов кипения жидкостей, не осложненных сопутствующими химическими процессами.
а)
б)
Рис. 10. Данные ТГ, ДТГ и ДСК: а – антраниловой кислоты (масса навески 13,6, 42, 47 и 48 мг), б – дииод- пара -ксилола (17,1 мг)
Можно отметить, что близкую к параболической зависимость измеренной высоты пика плавления (кристаллизации) меди в вакууме от скорости нагревания (рис. 11) [25] можно считать близким аналогом зависимости на рис. 4. В данном случае причина задержки также кроется в конечной теплопроводности среды между образцом и термопарой. При росте разницы температур (за счет роста скорости нагрева или за счет роста массы навески) скорость теплопередачи увеличивается и зависимость роста погрешности приближается к параболической.
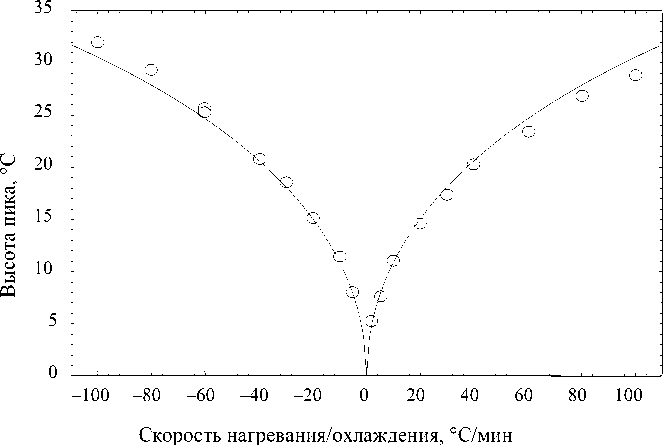
Рис. 11. Зависимость высоты пика плавления (кристаллизации) меди в вакууме от скорости ее нагревания/охлаждения
Температура окончания испарения, определенная по минимумам на кривых ДТГ, оказывается на 0,2–1,0 °С ниже, чем аналогично определенная по кривой ДСК. Это связано с тем, что весы имеют намного более короткое время отклика на изменения массы, по сравнению с откликом ДСК на окончание поглощения тепла образцом. Действительно, передача тепла между образцом и спаем термопары, находящимся сравнительно далеко от образца (они разделены стенкой корундового тигля, а также воздушным зазором между тиглем и термопарным столиком), а также требующееся время на прогрев тигля и термопарного столика обуславливают некоторую тепловую инерцию измерительной системы. Если температура окончания испарения, определенная по минимуму пика на кривой ДТГ, оказывается выше, чем аналогично определенная по кривой ДСК, то следует предполагать наличие примесей в образце или протекание побочных процессов (например, полимеризации или окисления).
Возможность использования кривых ДТГ для определения температур кипения требует отдельного рассмотрения и осторожности. Эти кривые, по сравнению с ДСК, обычно содержат больше «шума», в случае антраниловой кислоты, возможно, связанному с ее частичным разложением (декарбоксилированием) (см. рис. 10а). Сравнение трех методов обработки результатов синхронного термического анализа показывает преимущество предложенного метода (рис. 12).
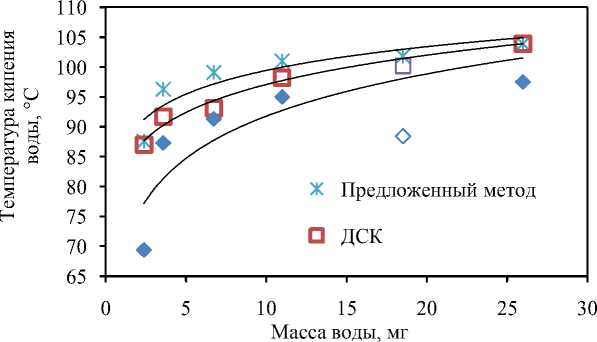
Рис. 12. Температуры кипения воды, определенные по кривым ДСК предложенным и стандартным методом (применяемым для определения начала пика плавления), а также по кривым ДТГ стандартным методом (для начала пика плавления)
В диапазоне масс навесок воды от 3,6 до 26 мг он имеет наименьшие отклонения от справочного значения (от 96,3 до 103,9 °С). Значения температур кипения, определенные стандартным методом (используемым для определения точки плавления), имеют намного более заметные отклонения в сторону низких температур (от 91,7 до 103,9 °С). Температуры кипения, определенные по кривым ДТГ методом, используемым для определения точки плавления, приводят к еще более низким значениям (от 69,4 до 97,5 °С).
Выводы
Для повышения точности определения температуры кипения следует пользоваться максимальной по массе навеской и такой обработкой кривой ДСК, при которой точку кипения вычисляют как точку пересечения касательной к базовой линии ДСК и линии, аппроксимирующей наклон фронта пика кипения, начинающейся вблизи вершины пика испарения. Критерием правильности построения касательной к линии фронта пика кипения является ее близость к наклону линии фронта пика плавления чистого индия, висмута или цинка. Чем выше масса жидкости, тем более высокие и точные результаты определения ее температуры кипения.
Теплота кипения рассчитывается из площади пика кипения по той же методике, что и теплота плавления. Точность определения теплоты кипения остается достаточно высокой вне зависимости от массы жидкости.
Исследование выполнено за счет гранта Российского научного фонда (проект №21-7320019).
Список литературы Об использовании синхронного термоанализатора Netzsch 449 Jupiter для определения точки кипения веществ
- Берг Г.Г. Введение в термографию. М.: Наука, 1969. 396 с.
- Уэндландт У. Термические методы анализа. Пер. с англ. под ред. В.А. Степанова и В.А. Берштейна. М.: Мир, 1978. 526 с.
- Шестак Я. Теория термического анализа: Физико-химические свойства твердых неорганических веществ. М.: Мир, 1987. 456 с.
- Альмяшев В.И. Термические методы анализа: учеб. пособие. СПб: Изд-во СПбГЭТУ (ЛЭТИ), 1999. 40 с.
- Термогравиметрия: учеб. пособие для студентов III курса химического факультета / сост. В.С. Корзанов, М.Г. Котомцева, Р.И. Юнусов. Пермь: Изд-во Перм. ун-та, 2007. 71 с.
- Goodrum J.W., Siesel E.M.Thermogravimetric analysis for boiling points and vapor pressure // Journal of Thermal Analysis. 1996. Vol. 46. Р. 1251-1258. DOI: 10.1007/BF01979239.
- Barrall II Edward M. Precise determination of melting and boiling points by differential thermal analysis and differential scanning calorimetry // Thermochimica Acta. 1973. Vol. 5. P. 377-389. DOI: 10.1016/0040-6031(73)80016-4.
- Goodrum J.W. Rapid Measurements of Boiling Point and Vapor Pressure of Short-Chain Triglycerides by Thermogravimetric Analysis // JAOCS. 1997. Vol. 74, no. 8. Р. 947-950. DOI: 10.1007/s11746-997-0009-0.
- Troni K.L., Damaseno D.S., Ceriani R. Evaluation of the variation of the differential scanning calorimetry method for measuring the boiling points of binary mixtures at subatmospheric pressure // J. Chem. Eng. Data. 2020. Vol. 65, no. 7. Р. 3334-3343. DOI: 10.1021/acsjced.0c00111.
- O'Connell C., Dollimore D. Determination of the melting point using derivative thermogravi-metry // Instrumentation Science & Technology. 1999. Vol. 27, no. 1. Р. 13-21. DOI: 10.1080/10739149908085826.
- Strella S. Differential Thermal Analysis of Polymers. II. Melting // Journal of applied polymer science. 1963. Vol. 7. P. 1281-1289. DOI: 10.1002/app.1963.070070409.
- Tubes R.K. Melting Point and Heat of Fusion of Poly(viny1 Alcohol) // Journal of polymer science: PART A. 1965. Vol. 3. P. 4181-4189. DOI: 10.1002/pol.1965.100031213.
- Fischer H. Calibration of micro-thermal analysis for the detection of glass transition temperatures and melting points. Repeatability and reproducibility / H. Fischer // Journal of Thermal Analysis and Calorimetry. 2008. Vol. 92, no. 2. Р. 625-630. DOI: 10.1007/s10973-007-8554-1.
- Reed T.B., Fahey R.E., Strauss A.J. Sealed crucible technique for thermal analysis of volatile compounds up to 2500 °C: melting points of EuO, EuS, EuSe and EuTe // Journal of Crystal Growth. 1972. Vol. 15. P. 174-178. DOI: 10.1016/0022-0248(72)90116-9.
- Kerr N.J., Landis P.S. Microboiling and Melting Point Determination by Differential Thermal Analysis // Analytical chemistry. 1968. Vol. 40, no. 7. Р. 1176-1176. DOI: 10.1021/ac60263a007.
- Ривкин С.Л., Александров A.A. Теплофизические свойства воды и водяного пара. М.: Энергия, 1980. 424 с.
- Химическая энциклопедия. Т. 2 / под ред. И.Л. Кнунянца. М.: Советская энциклопедия, 1990. 671 с.
- Химическая энциклопедия. Т. 5 / под ред. Н.С. Зефирова. М.: Советская энциклопедия, 1998. 783 с.
- Химическая энциклопедия. Т. 3 / под ред. И.Л. Кнунянца. М.: Советская энциклопедия, 1992. 639 с.
- Химическая энциклопедия. Т. 1 / под ред. И.Л. Кнунянца. М.: Советская энциклопедия, 1988. 623 с.
- Maki T., Takeda K. Benzoic Acid and Derivatives // Ullmann's Encyclopedia of Industrial Chemistry. 2000. DOI: 10.1002/14356007.a03_555.
- Halogen—halogen interactions in diiodo-xylenes / E. Albright, J. Cann, A. Decken, S. Eisler // CrystEngComm. 2017. Vol. 19. Р. 1024-1027. DOI: 10.1039/c6ce02339h.
- Stevens W.H., Pepper J.M., Lounsbury M. The decarboxylation of anthranilic acid // Canadian Journal of Chemistry. 2011. Vol. 30, no. 7. P. 529-540. DOI: 10.1139/v52-065.
- Triboluminescence-structure relationships in polymorphs of hexaphenylcarbodiphosphorane and anthranilic acid, molecular crystals, and salts / G.E. Hardy, W.C. Kaska, B.P. Chandra, J.I. Zink // Journal of the American Chemical Society. 1981. Vol. 103, no. 5. P. 1074-1079. DOI: 10.1021/ja00395a014.
- Жеребцов Д.А. Совершенствование методики высокотемпературного дифференциального термического анализа и определение некоторых термодинамических параметров систем CaO-Al2O3 и ZrO2-Al2O3: дис. ... канд. хим. наук. Челябинск, 2000. 126 с.
