Особенности электронной структуры сложных халькогенидов, галогенидов и оксидов, определенные по результатам квантово-механических расчетов

Автор: Лаврентьев А.А., Габрельян Б.В., Туан В.В., Калмыкова К.Ф.
Журнал: Advanced Engineering Research (Rostov-on-Don) @vestnik-donstu
Рубрика: Информатика, вычислительная техника и управление
Статья в выпуске: 1 т.26, 2026 года.
Бесплатный доступ
Введение. Современная квантовая и оптоэлектроника, нелинейная оптика предъявляют высокие требования к физико-химическим характеристикам используемых материалов. Это заставляет в том числе искать новые материалы, которые обладали бы свойствами, необходимыми в той или иной области применения. Но при таком подходе могут усложняться состав и кристаллическая структура полученных соединений. Электронная структура сложных соединений определяет их электрические, оптические, магнитные, химические свойства. Эти свойства являются индивидуальными для каждого соединения. Тем не менее, известно, что разные, но близкие по каким-то важным параметрам соединения, например изоэлектронные, обладают подобием в строении своих электронных оболочек. Накопление такой информации по отдельным соединениям и их группам приводит к необходимости обобщения полученных данных. И цель настоящей работы — рассмотреть некоторые общие характеристики электронной структуры, проявляемые группами разных соединений (халькогенидов, галогенидов и оксидов). Материалы и методы. Предметом изучения были три группы соединений: халькогениды Tl3TaS4, Tl3PS4, Sn2P2S6, InPS4, Cu2CdGeS4, Ag2CdSnS4, Ag2HgSnS4, галогениды Cs2HgX4 (X = Cl, Br, I), группа APb2Br5 (A = K, Rb) и оксиды La2Zr2O7, Nd2Zr2O7, Sm2Zr2O7, Eu2Zr2O7, Gd2Zr2O7. Метод исследования — квантово-механические расчеты в рамках теории функционала электронной плотности с различными обменно-корреляционными потенциалами. Использовались потенциалы, позволяющие учитывать сильные корреляции d- и f-электронов и получать значение ширины запрещенной зоны, близкое к экспериментальному. Результаты исследований. Проведены квантово-механические расчеты плотностей электронных состояний и оптических характеристик ряда халькогенидов, галогенидов и оксидов. Приведены парциальные и полные плотности электронных состояний (Densities of States — DOS). Выполнено сравнение полной плотности состояний с экспериментальными рентгеноэлектронными спектрами (X-ray photoelectron Spectra — XPS). Подтверждена адекватность результатов проведенных расчетов. Вершину валентной полосы формируют p-состояния наиболее электроотрицательных элементов (S, Se, Te, Br, O), в то время как дно валентной полосы образовано s-состояниями также электроотрицательных элементов. Обсуждение. По результатам проведенных расчетов сделаны обобщающие выводы о сходстве в строении валентной полосы рассмотренных соединений. На примере соединения Tl3TaS4 показано, что в твердом теле, по сравнению с энергиями в свободном атоме, для электроотрицательных элементов энергия связи уровней значительно уменьшается, а для электроположительных — увеличивается. Редкоземельный элемент (в качестве примера взят Eu2Zr2O7) вносит существенные дополнения в картину электронно-энергетического строения, так что электронные состояния редкоземельного элемента (4f-, 5p-) и 5s-состояния европия (Eu) изменяют строение валентной полосы пирохлора (Eu2Zr2O7). Рассчитанные в работе полные и парциальные плотности электронных состояний (DOS) сравнивались с экспериментальными рентгеновскими и рентгеноэлектроными (XPS) спектрами, которые подтвердили адекватность проведенных расчетов, при этом на рассчитанных кривых DOS имеются многочисленные элементы тонкой структуры, «замазанные» за счет аппаратурного искажения на экспериментальных кривых. Таким образом, расчет очень хорошо дополняет эксперимент, давая более детальную картину электронно-энергетического строения исследованных соединений. Заключение. Достигнута цель исследования — рассмотрены некоторые общие характеристики электронной структуры, проявляемые группами разных соединений (халькогенидов, галогенидов и оксидов). Решены задачи выявления состояний определяющих особенности электронной структуры и оптических характеристик исследованных групп соединений. Исследование может быть использовано при моделировании новых материалов с заданными свойствами.
Пирохлоры, электронно-энергетическая структура, метод функционала плотности, обменно-корреляционные потенциалы, оптические свойства
Короткий адрес: https://sciup.org/142247505
IDR: 142247505 | УДК: 538.915;538.958 | DOI: 10.23947/2687-1653-2026-26-1-2283
Electronic Structure Characteristics of Complex Chalcogenides, Halides, and Oxides from Quantum-Mechanical Calculations
Introduction. Modern quantum and optoelectronics, as well as nonlinear optics, place high demands on the physical and chemical properties of the materials used. This necessitates, among other things, the search for new materials that possess the properties required for a given application. At the same time, this approach can complicate the composition and crystal structure of the resulting compounds. The electronic structure of complex compounds determines their electrical, optical, magnetic, and chemical properties. These properties are unique to each compound. However, it is known that different compounds that are similar in some important parameters, for example isoelectronic ones, exhibit similarities in the structure of their electronic shells. The accumulation of such information on individual compounds and their groups necessitates generalizing the data obtained. The research objective is to consider some general characteristics of the electronic structure exhibited by groups of different compounds (chalcogenides, halides, and oxides). Materials and Methods. The subject of study was three groups of compounds: chalcogenides Tl3TaS4, Tl3PS4, Sn2P2S6, InPS4, Cu2CdGeS4, Ag2CdSnS4, Ag2HgSnS4, halides Cs2HgX4 (X = Cl, Br, I), group APb2Br5 (A = K, Rb), and oxides La2Zr2O7, Nd2Zr2O7, Sm2Zr2O7, Eu2Zr2O7, Gd2Zr2O7. The research method involved quantum-mechanical calculations within the framework of density functional theory with various exchange-correlation potentials. Potentials were used that allowed for strong correlations between d- and f-electrons and yield a band gap value close to the experimental value. Results. Quantum-mechanical calculations of the electronic state densities and optical characteristics of a number of chalcogenides, halides, and oxides were performed. Partial and total electron densities of states (DOS) were presented. The total density of states was compared with experimental X-ray photoelectron spectra (XPS). The validity of the calculation results was confirmed. The top of the valence band was formed by the p-states of the most electronegative elements (S, Se, Te, Br, O), whereas the bottom of the valence band was formed by the s-states of these same electronegative elements. Discussion. Based on the calculations, general conclusions were drawn regarding the similarities in the valence band structure of the compounds considered. Using the compound Tl3TaS4 as an example, it was shown that in a solid, compared to the energies in a free atom, the binding energy of the levels for electronegative elements was significantly reduced, while for electropositive elements, it was increased. A rare-earth element (using Eu2Zr2O7 as an example) significantly altered the electron-energy structure, such that the electron states of the rare-earth element (4f-, 5p-) and the 5s-states of europium (Eu) altered the structure of the valence band of pyrochlore (Eu2Zr2O7). The calculated total and partial DOS were compared with experimental X-ray and X-ray photoelectron spectra, which confirmed the accuracy of the calculations. However, the calculated DOS curves contained numerous fine-structure elements that were obscured by instrumental distortion in the experimental curves. Thus, the calculation complemented the experiment very well, providing a more detailed picture of the electron-energy structure of the studied compounds. Conclusion. The research objective was achieved: some general characteristics of the electronic structure exhibited by groups of different compounds (chalcogenides, halides, and oxides) were examined. The problems of identifying the states that determined the features of the electronic structure and optical characteristics of the studied groups of compounds were solved. This research can be used in the modeling of new materials with desired properties.
Текст научной статьи Особенности электронной структуры сложных халькогенидов, галогенидов и оксидов, определенные по результатам квантово-механических расчетов
Original Theoretical Research
Electronic Structure Characteristics of Complex Chalcogenides, Halides, and Oxides from Quantum-Mechanical Calculations
Anatoliy A. Lavrentyev 1 © в , Boris V. Gabrelian1 © , Vu Van Tuan2, 3 ©, Kseniya F. Kalmykova1
-
1 Don State Technical University, Russian Federation
-
2 Institute of Computational Science and Artificial Intelligence, Van Lang University, Ho Chi Minh City, Socialist Republic of Vietnam
-
3 School of Technology, Van Lang University, Ho Chi Minh City, Socialist Republic of Vietnam
Introduction . Modern quantum and optoelectronics, as well as nonlinear optics, place high demands on the physical and chemical properties of the materials used. This necessitates, among other things, the search for new materials that possess the properties required for a given application. At the same time, this approach can complicate the composition and crystal structure of the resulting compounds. The electronic structure of complex compounds determines their electrical, optical, magnetic, and chemical properties. These properties are unique to each compound. However, it is known that different compounds that are similar in some important parameters, for example isoelectronic ones, exhibit similarities in the structure of their electronic shells. The accumulation of such information on individual compounds and their groups necessitates generalizing the data obtained. The research objective is to consider some general characteristics of the electronic structure exhibited by groups of different compounds (chalcogenides, halides, and oxides).
Materials and Methods. The subject of study was three groups of compounds: chalcogenides Tl 3 TaS 4 , Tl 3 PS 4 , Sn 2 P 2 S 6 , InPS 4 , Cu 2 CdGeS 4 , Ag 2 CdSnS 4 , Ag 2 HgSnS 4 , halides Cs 2 HgX 4 (X = Cl, Br, I), group APb 2 Br 5 (A = K, Rb), and oxides La 2 Zr 2 O 7 , Nd 2 Zr 2 O 7 , Sm 2 Zr 2 O 7 , Eu 2 Zr 2 O 7 , Gd 2 Zr 2 O 7 . The research method involved quantum-mechanical calculations within the framework of density functional theory with various exchange-correlation potentials. Potentials were used that allowed for strong correlations between d- and f-electrons and yield a band gap value close to the experimental value.
Results . Quantum-mechanical calculations of the electronic state densities and optical characteristics of a number of chalcogenides, halides, and oxides were performed. Partial and total electron densities of states (DOS) were presented. The total density of states was compared with experimental X-ray photoelectron spectra (XPS). The validity of the calculation results was confirmed. The top of the valence band was formed by the p-states of the most electronegative elements (S, Se, Te, Br, O), whereas the bottom of the valence band was formed by the s-states of these same electronegative elements.
Discussion. Based on the calculations, general conclusions were drawn regarding the similarities in the valence band structure of the compounds considered. Using the compound Tl 3 TaS 4 as an example, it was shown that in a solid, compared to the energies in a free atom, the binding energy of the levels for electronegative elements was significantly reduced, while for electropositive elements, it was increased. A rare-earth element (using Eu 2 Zr 2 O 7 as an example) significantly altered the electron-energy structure, such that the electron states of the rare-earth element (4f-, 5p-) and the 5s-states of europium (Eu) altered the structure of the valence band of pyrochlore (Eu 2 Zr 2 O 7 ). The calculated total and partial DOS were compared with experimental X-ray and X-ray photoelectron spectra, which confirmed the accuracy of the calculations. However, the calculated DOS curves contained numerous fine-structure elements that were obscured by instrumental distortion in the experimental curves. Thus, the calculation complemented the experiment very well, providing a more detailed picture of the electron-energy structure of the studied compounds.
Conclusion. The research objective was achieved: some general characteristics of the electronic structure exhibited by groups of different compounds (chalcogenides, halides, and oxides) were examined. The problems of identifying the states that determined the features of the electronic structure and optical characteristics of the studied groups of compounds were solved. This research can be used in the modeling of new materials with desired properties.
Введение. В задачи полупроводникового материаловедения входит получение на основе квантово-механических расчетов и привлечения экспериментальных рентгеновских и рентгеноэлектронных спектров достоверной информации об электронно-энергетической структуре и химической связи, а также об оптических характеристиках сложных полупроводников. Преимуществом применения расчетных моделей является возможность проведения исследований гипотетических, еще не синтезированных [1] соединений, а также отсутствие необходимости в создании подходящих образцов до начала исследований. Целью настоящей работы является обобщение результатов изучения разных групп соединений с помощью надежных, имеющих широкую апробацию вычислительных моделей. В расчетах использовались различные обменно-корреляционные потенциалы, но результаты, наиболее близкие к экспериментальным данным, были ранее получены с модифицированным Tran and Blaha потенциалом Beke-Johnsona (modified Becke-Johnson — mBJ), с добавлением Хаббардовской U-поправки на сильное взаимодействие электронов в d- и f-оболочках, а также с учетом спин-орбитального взаимодействия (Spin-Orbit Coupling — SOC), особенно для оболочек тяжелых элементов. Все ссылки на оригинальные статьи авторов сделанных приближений в расчетах можно найти в работах [1 –4] .
По всем исследованным соединениям проведен полномасштабный литературный обзор свойств и всех исследований электронной структуры как экспериментальными, так и теоретическими расчетными методами. Обзор показал следующее. Соединение Tl 3 TaS 4 относится к материалам, используемым в устройствах, основанных на применении поверхностных акустических волн (ПАВ) [5] , и находящим широкое применение в нелинейной оптике и устройствах связи, таких как мобильная телефонная и телевизионная связь [6] . Для удовлетворения современных технологических требований исследования по совершенствованию ПАВ-материалов должны продолжаться и дальше. Ранее было проведено несколько экспериментов с целью изучения края оптического поглощения, электронной структуры, особенностей химической связи в Tl 3 TaS 4 [7] . Однако на точность получаемых экспериментальных значений оптической запрещенной щели, коэффициента поглощения существенно влияют трудности наблюдения d-орбиталей в широком энергетическом интервале и ряд внешних факторов [8] . Электронная структура, а также оптические свойства Tl 3 TaS 4 могут быть подробно изучены на основе ab initio расчетов, которые интенсивно развивались и доказали свою способность воспроизводить достоверные свойства материалов [9] . Соединение Eu 2 Zr 2 O 7 относится к группе пирохлоров с общей формулой A 2 B 2 O 7 , где A и B — металлические катионы, которые могут быть трехвалентными (Eu) и четырехвалентными (Zr) [10] . Пирохлоры обладают значительной диэлектрической проницательностью, проявляют уникальные магнитные [11] , химические, механические, электрические свойства [12] . Благодаря этому их можно использовать как керамические покрытия термобарьеров, газовых сенсоров, металлооксидных транзисторов, твердых электролитов в токсичных элементах [13] , как иммобилизаци-онных носителей актиноидов в ядерных отходах и катализаторов окислительных реакций [14] . Для создания более эффективных, надежных и функциональных устройств на основе пирохлоров [15] проводились исследования с учетом развития новых технологий в области лазерной техники, оптики и материаловедения [15] . Расчеты электронноэнергетической структуры различных пирохлоров велись и в рамках теории функционала плотности [16] ,
Информатика, вычислительная техника и управление
с использованием обменно-корреляционного потенциала в приближении локальной плотности (LDA) и обобщенного градиентного приближения (GGA). В [16] отмечалось важность учета поправки Хаббарда U при расчете энергии d- и f-состояний. Однако получить в расчетах значения ширины запрещенной щели E g , сравнимые с экспериментальными, не удалось (расчетные значения занижены, по сравнению с экспериментальными).
Чтобы получить достоверную картину электронно-энергетических полос в исследуемом пирохлоре Eu 2 Zr 2 O 7 , а затем рассчитать оптические характеристики этого кристалла, потребовалось использовать другие приближения к обменно-корреляционному потенциалу.
Таким образом, судя по данным представленной научной литературы, расчетов было проведено много, но они требуют улучшения используемых приближений (особенно это касается обменно-корреляционных потенциалов), вот почему в ранних работах авторами использовался потенциал Беке-Джонсона (mBJ). Кроме того, в вышеуказанных публикациях приведены для сравнения с расчётными кривыми и экспериментальные кривые рентгеновских и ренгеноэлектронных спектров, полученных авторами [1] и соавторами [2 –4] . Цель настоящей работы — рассмотреть характеристики электронной структуры, проявляемые группами разных соединений (халькогенидов, галогенидов и оксидов).
Материалы и методы. На основе полнопотенциального, полноэлектронного метода присоединенных плоских волн (Full Potential Linearized Augmented Plane Waves — FPLAPW), реализованного в программном комплексе Wien2k [17] , ранее были проведены модельные квантово-механические расчеты электронно-энергетического строения следующих трех групп полупроводниковых соединений:
-
- халькогенидов Tl 3 TaS 4 [1] , Tl 3 PS 4 , Sn 2 P 2 S 6 , InPS 4 , Cu 2 CdGeS 4 , Ag 2 CdSnS 4 , Ag 2 HgSnS 4 [2] ;
-
- галогенидов Cs 2 HgX 4 (X — Cl, Br, I), группа APb 2 Br 5 (A — K, Rb) [2] ;
-
- оксидов La 2 Zr 2 O 7 и Nd 2 Zr 2 O 7 [3] ; Ln 2 Zr 2 O 7 (Ln = La, Nd, Sm, Eu, Gd) [4] .
В элементарной ячейке кристалла каждый атом окружался muffin-tin сферой (mt-сфера), в результате чего весь ее объем разбивался на области, занятые mt-сферами, и остальное междусферное пространство, а затем рассчитывался кристаллический потенциал как в mt–сферах, так и в междусферье. Междусферный потенциал рассчитывался по методике, описанной в [18] .
Для атомов соединения использовались следующие mt-радиусы: R E m u t = 2,24 a.u.; R Z m r t = 1,96 a.u.; R m O t = 1,77 a.u. (a.u. — атомная единица длины).
Расчет обменно-корреляционного потенциала проводился в приближении GGA-PBE [19] или в приближении mBJ (модифицированный потенциал Беке-Джонсона [20] ). В соединении Eu 2 Zr 2 O 7 редкоземельный элемент Eu имеет недостроенную 4f7-оболочку, и для учета сильного кулоновского взаимодействия 4f-электронов на одном узле использовался Хаббардовский параметр U, что привело к обменно-корреляционному потенциалу PBE + U [21] и mBJ + U [22] . Как и для других соединений с 4f-оболочкой, расчет ЭЭС в Eu 2 Zr 2 O 7 был спин-поляризованным.
Электронная структура соединений с валентными s-, p-, d-электронами обсуждался в дальнейшем на примере соединения Tl 3 TaS 4 , кристаллизующегося в кубической структуре с пространственной группой I-43m, и значением параметра решетки a = 7,67 Å [1] .
Кристаллическая структура всех исследованных пирохлоров Ln 2 Zr 2 O 7 (Ln = La, Nd, Sm, Eu, Gd), соединений с валентными f-электронами одинакова и принадлежит кубической решетке с пространственной группой Fd3m. В расчетах Eu 2 Zr 2 O 7 использовалось значение параметра решетки a = 10,5438 Å, координаты атомов [4] приведены в таблице 1.
Координаты атомов в элементарной ячейке соединения Eu 2 Zr 2 O 7
Таблица 1
|
Атом |
Символы Вайкоффа |
x/a |
y/a |
z/a |
|
63 Eu |
16 d |
0,5 |
0,5 |
0,5 |
|
40 Zr |
16 c |
0 |
0 |
0 |
|
8 O1 |
48 f |
x = 0,33888 |
0,125 |
0,125 |
|
8 O2 |
8 b |
0,375 |
0,375 |
0,375 |
На рис. 1 показана кристаллическая структура и окружение атомов в пирохлоре Eu 2 Zr 2 O 7 [4] .
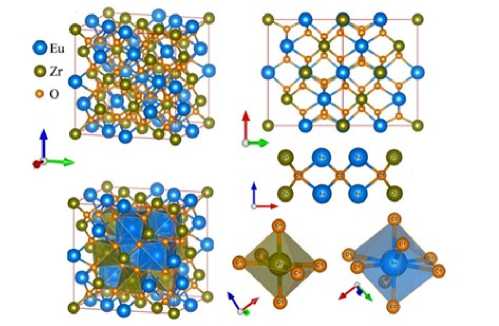
Рис. 1. Кристаллическая структура и ближайшее окружение атомов в пирохлоре Eu 2 Zr 2 O 7 . Расстояния между атомами: Eu-O1 = 2,5224 Å, Eu-O2 = 2,2828 Å, Zr-O1 = 2,0858 Å
По занятым электронным состояниям в валентной полосе и свободным электронным состоянием в полосе проводимости можно рассчитать комбинированную плотность состояний
J ( Й ю ) = 2 N ( E ') N ( E ' + Й ю ) dE', (1)
и далее, используя матричные элементы перехода из валентной полосы в полосу проводимости (рассматриваются только прямые дипольные переходы с Δ l = 1 соответствующих состояний отдельного атома, перекрестные переходы, как маловероятные, игнорируются), можно рассчитать мнимую часть тензора диэлектрической проницаемости [23] .
e 2 (ю) =
4п2 e 2 Q m 2ю2
x
^ кПп c| p,\kn 'c} (kn Ъ| pj\kn c^ fn ( 1 - f to') 3 ( E kn■ - E , - Й ю ) ,
nn G
где Ekn — собственная энергия системы с кристаллическим импульсом k и спином σ; m и e — масса и заряд электрона соответственно; V, p , | knpV) и fkn — объем элементарной ячейки, оператор импульса, волновая функция кристалла, функция распределения Ферми.
Действительная часть е 1 (ш) диэлектрической функции рассчитывалась по формуле Крамерса-Кронига:
/ х 2 ю^2 ( ю' )
e1 (ю) = 1 + — P d ю, п ю 2 -ю2
где P — главное значение интеграла.
Коэффициент поглощения a (ω), показатель преломления n (ω), коэффициент экстинкции k (ω), оптический коэффициент отражения R (ω) и спектр энергетических потерь электрона L (ω) выводятся из мнимой ε 2 (ω) и действительной ε 1 (ω) частей тензора диэлектрической проницаемости и рассчитываются, соответственно, по следующим формулам [23] .
Коэффициент поглощения:
. 2 ю k i (ю)
aij (ю)=(4)
Показатель преломления:
1 22
n‘J (ю) = -^= ^e,J (ю) +е2 (ю) +6/ (ю) .(5)
Коэффициент экстинкции:
1 2212
k(ю) = 72Ne (ю) + e2(ю) -eij (ю)] .
Оптический коэффициент отражения:
Rii ( ю ) =
(ni -1 ) 2 + kj 2 (n, J +1 ) 2 + kj
4 e J + i e 2 -1 7e j + i e iJ +1
Функция энергетических потерь:
L ( ю ) = - Im ( e 1 ) J =
e 2 (ю) e j ( ю ) 2 +e 2 ( ю ) 2
Информатика, вычислительная техника и управление
Результаты исследования. Электронная структура соединений с валентными s-, p-, d-состояниями на примере Tl 3 TaS 4 . Рассмотрены результаты исследования электронной энергетической структуры (ЭЭС) в соединении Tl 3 TaS 4 из работы [1] , затем сделаны некоторые обобщения по проведенным исследованиям ЭЭС вышеуказанных групп халькогенидов, галогенидов и оксидов.
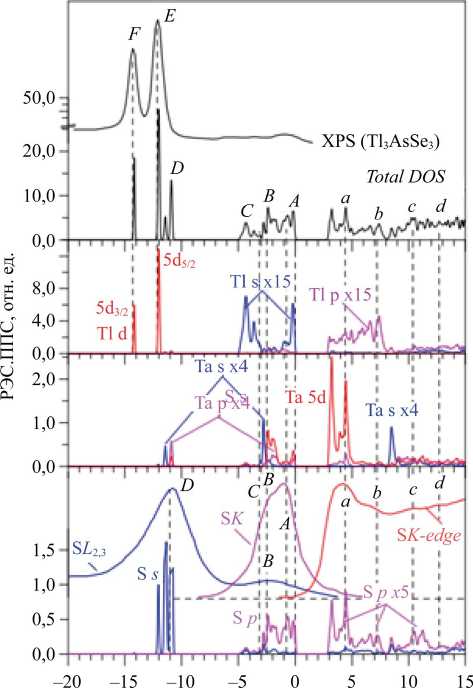
На рис. 2 показано сравнение рассчитанных полной и парциальных плотностей электронных состояний с рентгеновским K- и L 2,3 -спектрами эмиссии и SK-спектром поглощения.
Энергия, эВ
Рис. 2. Полная и парциальные плотности состояний Tl 3 TaS 4 , рассчитанные методом mBJ+U+SOC, в сравнении с экспериментальными XPS, рентгеновскими эмиссионными S K в1,3 и S £2,3 спектрами и рентгеновским спектром S K -поглощения
Рассчитанное в приближении mBJ+U+SOC значение ширины запрещенной щели Eg = 2,842 эВ близко к экспериментальному значению Eg = 2,7 эВ [24] .
Исходя из положения по энергии главных максимумов рентгеновских эмиссионных спектров (SK- и SL 2,3 -полос) и максимумов рассчитанных плотностей электронных состояний в полупроводниковом соединении Tl 3 TaS 4 (рис. 2), была построена энергетическая диаграмма максимумов основных энергетических полос этого соединения (рис. 3) в сравнении с энергетическими уровнями в свободном атоме из работы [25] , причем отсчет последних велся от вакуумного нуля. Вакуумный нуль отделен от нуля, соответствующего вершине валентной полосы ( Ev ) в Tl 3 TaS 4 , величиной работы выхода (φ) и половиной ширины запрещенной полосы ( Eg ). Для родственных полупроводников найдены следующие значения работы выхода: для TlAsS 4 и Tl 3 AsS 3 φ = 5,5 эВ, для Tl 3 AsS 4 φ = 5,5 эВ [26] , что позволило предположить значение φ = 5,5 эВ и для Tl 3 TaS 4 .
Как видно на рис. 3, наблюдается симбатное уменьшение энергии связи валентных 3p- и 3s-уровней серы, наиболее электроотрицательного (ЭО) элемента соединения Tl 3 TaS 4 (ЭО = 2,44). В свободном атоме энергии связи 3p- и 3s- уровней равны –10,36 и –20,20 эВ соответственно (таблица 2), в то время как в кристалле Tl 3 TaS 4 средние значения энергий максимумов полос этих состояний равны для 3p-состояний примерно –1,5÷–2 эВ и для 3s-состояний -11^12 эВ.
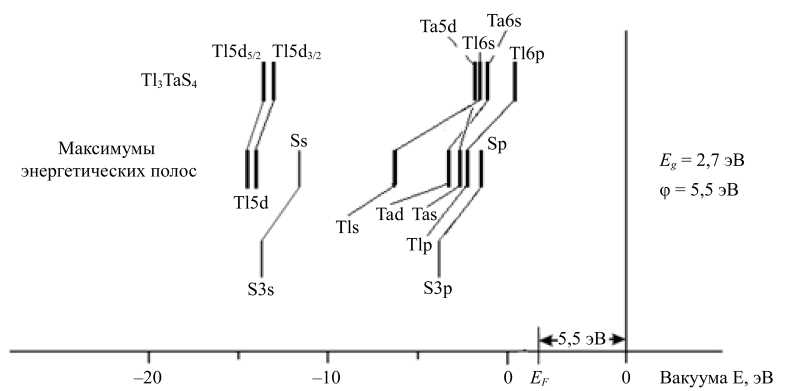
Рис. 3. Энергетические уровни в свободном атоме [25] и максимумы экспериментальных энергетических полос в полупроводниковом соединении Tl 3 TaS 4 относительно вакуумного нуля. Нуль на шкале энергии соответствует вершине валентной полосы ( E v ), E F — энергия Ферми, взятая посередине запрещенной щели и на расстоянии работы выхода от вакуумного нуля
В твердом теле электронная плотность оттягивается к более электроотрицательному атому (в данном случае к сере S), что приводит к увеличению экранировки ядра этого атома, а значит, как следствие, к понижению энергии связи как 3p-уровней, так и 3s-уровней, по сравнению с их значениями в свободном атоме (таблица 2).
Энергии связи валентных и полуостовных электронов в свободных атомах, входящих в соединение Tl 3 TaS 4 (в эВ) [25]
Таблица 2
|
16 S |
3s2 M 1 |
3p4 M 2 |
||
|
20,20 |
10,36 |
|||
|
73 Ta |
5d3 O 4 |
5s2 P 1 |
||
|
8,3 |
7,9 |
|||
|
81 Tl |
5d 10 O 4 — O 5 |
6s2 P 1 |
6p1 P 2 |
|
|
21 — 19 |
8 |
6,11 |
||
Как указано в монографии Блохина [27] , эффективный заряд ядер определяется не только внутренними, но и внешними по отношению к данной оболочке электронами, т. е. всеми электронами атома. Согласно этим представлениям, Z эфф = Z – σ 1 , где σ 1 — постоянная полного экранирования. Определение постоянной экранирования не входило в задачи настоящей работы и является весьма сложной теоретической задачей.
Для иллюстрации на рис. 4 приведено схематическое изображение сдвигов внутренних уровней для положительного и отрицательного ионов.
Изолированный атом
Положительный Отрицательный
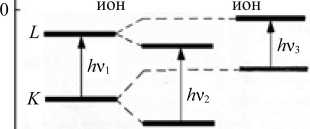
Рис. 4. Схематическое изображение химических сдвигов внутренних уровней для положительного и отрицательного ионов, по сравнению с изолированными атомами: h ν 3 < h ν 1 < h ν 2
Информатика, вычислительная техника и управление
Действительно, у более электроположительных элементов в Tl 3 TaS 4 , а именно Tl и Ta, энергии связи их валентных уровней увеличиваются, по сравнению с энергиями в свободном атоме (рис. 3 и таблица 2). Экранировка у Tl и Tа становится меньше за счет оттягивания электронной плотности к атому S, и это приводит к увеличению энергии связи валентных и полувалентных уровней металлов.
Подобные выводы о поведении электронных состояний можно сделать по всем исследованным соединениям (результаты опубликованы в работах [1 –4] ).
Таким образом, электронные p- состояния наиболее электроотрицательных атомов (S, Se, Te, Br, O) в исследуемых халькогенидах, галогенидах и оксидах формируют верхнюю часть валентной полосы, что связано с существенным уменьшением энергии связи этих состояний, по сравнению с их значениями в свободном атоме. Такое уменьшение энергии связи p- состояний можно объяснить перетеканием электронной плотности к более электроотрицательному атому, а значит, и увеличению экранировки этих состояний от ядра. Электронные s- состояния наиболее электроотрицательных атомов (S, Se, Te, Br, O) в халькогенидах, галогенидах и оксидах образуют дно валентной полосы, и их энергия связи также уменьшается, по сравнению с энергией в свободном атоме.
Электронная структура соединений с валентными f-состояниями на примере пирохлора Eu 2 Zr 2 O 7 . Чтобы сделать обобщающие выводы по электронно-энергетической структуре и оптическим характеристикам пирохлоров Ln 2 Zr 2 O 7 (Ln = La, Nd, Sm, Eu, Gd) [3, 4] , было рассмотрено соединение Eu 2 Zr 2 O 7 .
Валентные конфигурации элементов, входящих в соединение Eu 2 Zr 2 O 7, следующие:
Eu — 4f75s25p66s2
Zr — 4s24p65d25s2
O — 2s22p4.
На рис. 5 приведены полные плотности электронных состояний со спином вверх (spin-up) и спином вниз (spindown) для двух разных приближений: в приближении GGA–PBE+U и GGA–PBE+U+SOC с U = 5 эВ для 4f-со-стояний Eu. Добавление спин-орбитального взаимодействия (SOC) приводит к расщеплению 5p6-состояний Eu на 5p 1/2 и 5p 3/2 -состояния, а также к расщеплению 4p6-состояний Zr на 4p 1/2 и 4p 3/2 -состояния.
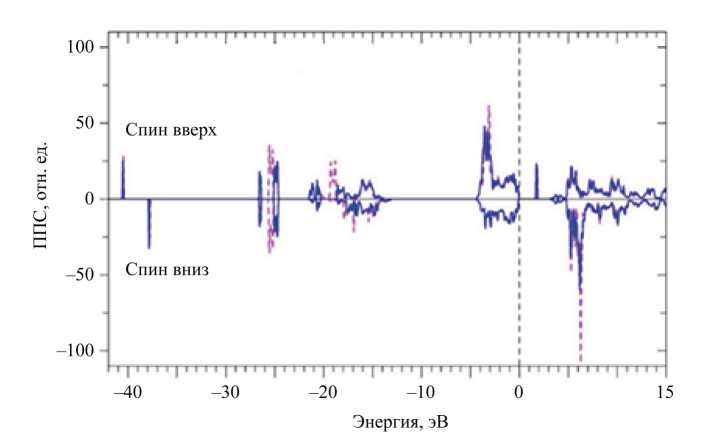
Рис. 5. Рассчитанные полные плотности электронных состояний со спином вверх (spin-up) и спином вниз (spin-down): пунктир (-----) — расчет в приближении GGA-PBE+U; сплошная линия — расчет в приближении GGA-PBE+U+SOC;
U = 5 эВ для 4f- состояний Eu. Нуль на шкале энергии соответствует вершине валентной полосы E v
По полным плотностям электронных состояний была проведена оценка ширины запрещенной полосы в Eu 2 Zr 2 O 7 , данные приведены в таблице 3.
Таблица 3
Ширина запрещенных полос E g для разных приближений расчета со спином вверх и спином вниз в Eu 2 Zr 2 O 7
|
Приближение |
Eg , эВ |
|
|
спин вверх |
спин вниз |
|
|
GGA-PBE |
0 |
0 |
|
GGA-PBE+U |
1,705 |
3,219 |
|
GGA-PBE+U+SOC |
1,667 |
|
Значение U для 4f-состояний Eu равно 5 эВ.
Отличие полной плотности электронных состояний со спином вверх и спином вниз на рис. 5 можно объяснить, используя парциальные плотности электронных состояний со спином вверх (рис. 6) и спином вниз (рис. 7)
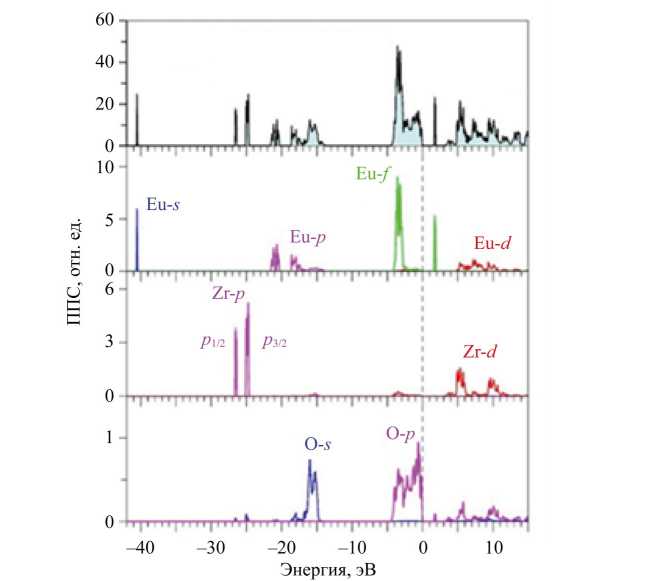
Рис. 6. Рассчитанные в приближении GGA-PBE+U+SOC полная и парциальные плотности электронных состояний для спина вверх (spin-up) в Eu 2 Zr 2 O 7 . Нуль шкалы энергии соответствует вершине валентной полосы E V
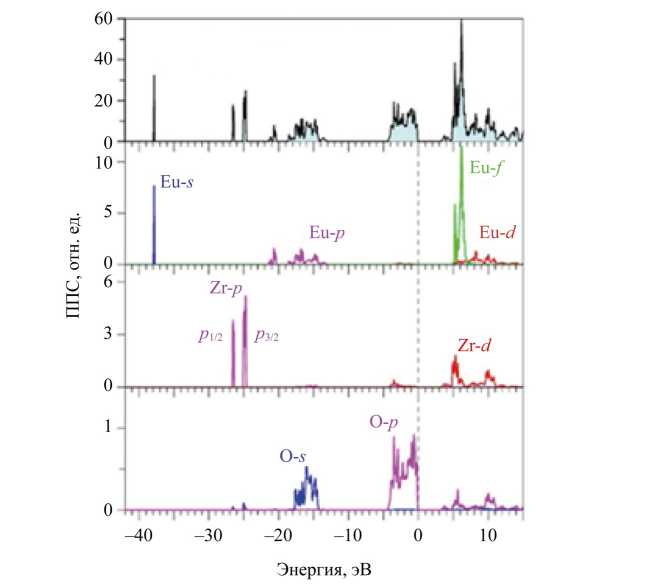
Рис. 7. Рассчитанные в приближении GGA-PBE+U+SOC полная и парциальные плотности электронных состояний для спина вниз (spin-down) в Eu 2 Zr 2 O 7 . Нуль ширины энергии соответствует вершине валентной полосы E V
Как видно на рис. 6 и 7, верхнюю часть валентной полосы Eu 2 Zr 2 O 7 формируют в основном 2p-состояния кислорода (область от 0 до 4,5 эВ). Некоторую примесь к 2p-состояниям O дают 4d- и 5s-состояния Zr, а также 6s-состояния редкоземельного элемента Eu. Наиболее значительной примесью в верхней части валентной полосы являются 4f-состояния Eu (область от –2 до –4 эВ). В недостроенной 4f-оболочке Eu имеется 7 электронов, спины которых выстраиваются по правилу Хунда в одном направлении: в данном расчете это спин вверх.
Информатика, вычислительная техника и управление
Спин-орбитальное расщепление (SOC) 5p-состояний Eu можно видеть и на парциальных плотностях со спином вверх и спином вниз (рис. 6 и 7). Поскольку 5p-состояния Eu углублены по отношению к 2s-состояниям О, то взаимодействие этих состояний уже не такое значительное, по сравнению с другими пирохлорами (La 2 Zr 2 O 7 , Nd 2 Zr 2 O 7 , Sm 2 Zr 2 O 7 ).
Незанятые 4f-состояния Eu появляются в настоящем расчете в полосе проводимости и со спином вверх (узкий пик на 2 эВ) и со спином вниз (узкий пик в области от 5 до 7 эВ).
Следует отметить еще одну важную, отличную от предыдущих пирохлоров особенность — пик 4f-состояний Eu со спином вверх расщепился на два небольших пика, что связано со спин-орбитальным взаимодействием 4f7-электронов. Это 4f 5/2 - и 4f 7/2 -состояния. У пирохлоров Nd 2 Zr 2 O 7 и Sm 2 Zr 2 O 7 этого не наблюдается.
Несмотря на то, что 4f-оболочка находится внутри 5s, 5p и 6s-оболочек, т. е. является внутренней оболочкой в атоме редкоземельного элемента (Nd, Sm, Eu, Gd), по энергии 4f-состояния в твердом теле, по данным настоящих расчетов, попадают в верхнюю часть валентной полосы, причем выше находятся только 6s-состояния. Объяснить такое энергетическое положение 4f-состояний можно значительным «центробежным» вкладом в потенциальную энергию: V ( r ) + l ( l + 1)Й 2 /2 mr , поскольку у 4f-электронов самое большое значение орбитального квантового числа ( l = 3).
Глубже по энергии находятся 2s-состояния О (область энергии от –14,5 до –18,5 эВ). Еще глубже по энергии находятся Eu 5p-состояния. Уже в атоме заполненная 5р-подоболочка Eu расщепляется за счет спин-орбиталь-ного взаимодействия (SOC) на 5p 1/2 (терм О 2 ) с энергией 30 эВ и на 5p 3/2 (терм О3) с энергией 26 эВ, что отражено в таблице 2. Спин-орбитальное расщепление (SOC) 5р-состояний Еu можно видеть и на парциальных плотностях со спином вверх и спином вниз (рис. 6 и 7). Поскольку 5р-состояния углублены по отношению к 2s-состояниям О, то взаимодействие этих состояний уже не такое значительное, по сравнению с предыдущими пирохлорами (La 2 Zr 2 O 7 , Nd 2 Zr 2 O 7 , Sm 2 Zr 2 O 7 ). Однако и в случае Eu 2 Zr 2 O 7 можно отметить примешивание 2s-состояний кислорода к 5p-состояниям редкоземельного элемента Eu, что связано c ковалентностью химсвязи Eu-O2.
В области энергий 25–27 эВ расположены расщепившиеся из-за спин-орбитального взаимодействия 4р6-со-стояния Zr. Эти состояния можно считать уже полуостовными, не участвующими в химической связи Zr и O1. Полуостовные 5s-состояния Eu оказываются самыми глубоколежащими из рассчитанных в настоящей работе валентных состояний. По правилу Паули два 5s-электрона Еu имеют разное направление спина, что приводит и к различию по энергии этих 5s-состояний Еu из-за действия на них магнитного поля 4f7-электронов Еu. 5s-электрон Еu со спином вверх углубляется до –40,5 эВ, а со спином вниз имеет энергию –38 эВ. Энергетическое расщепление для этих состояний 5s-электронов ∆E = 2,5 эВ.
Проведенные расчеты показали, что электронные состояния 5s-симметрии со спином вверх и со спином вниз расщепляются по энергии, что можно объяснить действием внутреннего магнитного поля 4f-электронов, спины которых выстраиваются по правилу Хунда и действуют на 5s-электроны со спином вверх и спином вниз (эффект Зеемана).
Увеличение числа 4f-электронов от 4 в Nd до 6 в Sm и до 7 в Eu (Nd 2 Zn 2 O 7 →Sm 2 Zr 2 O 7 →Eu 2 Zr 2 O 7 ) приводит к увеличению магнитного поля в 4f-оболочке и, как следствие, к увеличению расщепления 5s-состояний редкоземельного элемента со спином вверх и спином вниз (таблица 4).
Таблица 4
Величина расщепления Δ E 5s-состояний редкоземельного элемента со спином вверх и со спином вниз
Nd2Zn2O7 Sm2Zr2O7 Eu2Zr2O7 AE = 1,5 эВ A E = 2,5 эВ A E = 3,3 эВ
На рис. 8 приведены полные плотности электронных состояний со спином вверх и спином вниз в сравнении с экспериментальным рентгеноэлектронным спектром (XPS). Все особенности тонкой структуры XPS хорошо отражены на рассчитанных теоретически плотностях электронных состояний. Верхняя часть валентной полосы от 0 до –5 эВ формируется 2р-состояниями кислорода, что отражено на теоретической и экспериментальной кривых.
Наиболее интенсивный пик А на XPS соответствует 4f-состояниям Еu, поскольку сечение фотононизации 4f-состояний значительно превосходит сечение фотононизации 2р-состояний кислорода. На энергии ≈ –4 эВ находится интенсивный пик, как раз и отражающий 4f-состояния Eu.
Пологий пик B на XPS соответствует в основном 2ѕ-состояниям кислорода, а также небольшой доле 5p 3/2 -состояний Eu. Пик С на ХРЅ в основном образован 5p 1/2 -состояниями Еu с небольшой примесью 2ѕ-состояний кислорода. Пик D на XPS соответствует расщеплению за счет спин-орбитального взаимодействия (SOС) 4р-со-стояний Zr. Пик D на ХРЅ имеет явную асимметрию, что как раз и связано со спин-орбитальным расщеплением 4р6-электронов на 4р 1/2 и 4р 3/2 -состояния.
И, наконец, широкий пологий горб Е на рентгеноэлектронном спектре (ХРЅ) образован 5s-состояниями Еu, у которых происходит расщепление электронных состояний со спином вверх и спином вниз, «замазанное» на спектре из-за аппаратного искажения.
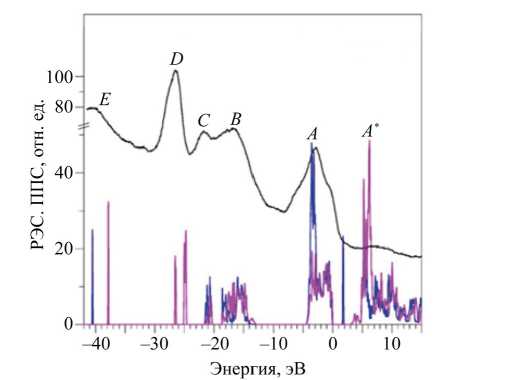
Рис. 8. Рассчитанные в приближении GGA-PBE+U+SOC полные плотности электронных состояний со спином вверх и спином вниз, в сравнении с экспериментальным рентгеноэлектронным спектром (XPS). Нуль на шкале энергии соответствует вершине валентной полосы Е v
Расчеты оптических характеристик на примере Eu 2 Zr 2 O 7 . Выходные данные расчетов ЭЭС соединения Eu 2 Zr 2 O 7 , а именно дисперсионные кривые E ( ⃗k ) и плотность электронных состояний (DOS), использовались для вычисления частотно-зависимой комплексной диэлектрической функции Ɛ(ω) = Ɛ 1 (ω) + iƐ 2 (ω). На первом этапе расчета диэлектрической функции вычислялась мнимая часть тензора диэлектрической функции Ɛ 2 (ω) по формуле (2). На рис. 9 приведена мнимая часть диэлектрической функции Ɛ 2 (ω) в зависимости от энергии фотона (частоты). Указаны основные пики и детали тонкой структуры кривой Ɛ 2 (ω): A, B, C, D, E, F, энергии которых даны в таблице 5.
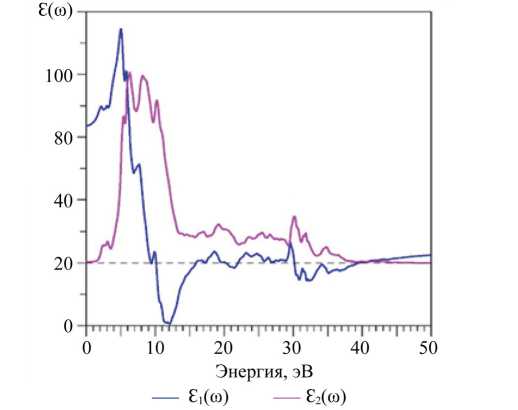
Рис. 9. Рассчитанная мнимая (ε 2 ) и действительная (ε 1 ) части диэлектрической проницаемости в Eu 2 Zr 2 O 7
Вместе с Ɛ 2 (ω) на рис. 9 приводится график действительной части диэлектрической проницаемости Ɛ 1 (ω), рассчитанной по формуле Крамерса–Кронига (3).
Таблица 5
Энергии выделенных пиков на мнимой части диэлектрической проницаемости Ɛ 2 (ω) (рис. 9), а также коэффициент отражения R (0), показатель преломления (0) в начале отсчета энергий
|
А |
Б |
В |
Г |
Д |
Е |
|
5,02 эВ |
8,25 |
10,16 |
19,22 |
25,62 |
30,16 |
|
Ɛ 1 (ω) |
8,726 |
Информатика, вычислительная техника и управление
n (0) = 2,954
R (0) = 24,423 %

Как и в других исследуемых пирохлорах, в расчете Ɛ 2 (ω) для Eu 2 Zr 2 O 7 перекрестные переходы между атомами не учитывались. Таким образом, можно проинтерпретировать основные особенности тонкой структуры мнимой части диэлектрической проницаемости Ɛ 2 (ω) следующим образом:
|
А(5,02 эВ) |
O |
p→s |
|
А’(6,5-7 эВ) |
Eu |
4f→d |
|
Б(8,25 эВ) |
O |
p→s |
|
В(10,16 эВ) |
O |
p→s |
|
Г(19,22 эВ) |
O |
s→p |
|
Д(25,62 эВ) |
Eu |
p→s |
|
Е(30,16 эВ) |
Zr |
p→s |
|
Ж(36 эВ) |
Eu |
p→s |
Для соединения Eu 2 Zr 2 O 7 по формулам (4–8) [23] рассчитывались, соответственно, коэффициент поглощения α(ω) (4), показатель преломления n (ω) (5), коэффициент поглощения k (ω) (6), оптический коэффициент отражения R (ω) (7) и спектр энергетических потерь электрона L (ω) (8). Вышеуказанные оптические характеристики для Eu 2 Zr 2 O 7 приведены на рис. 10, 11.
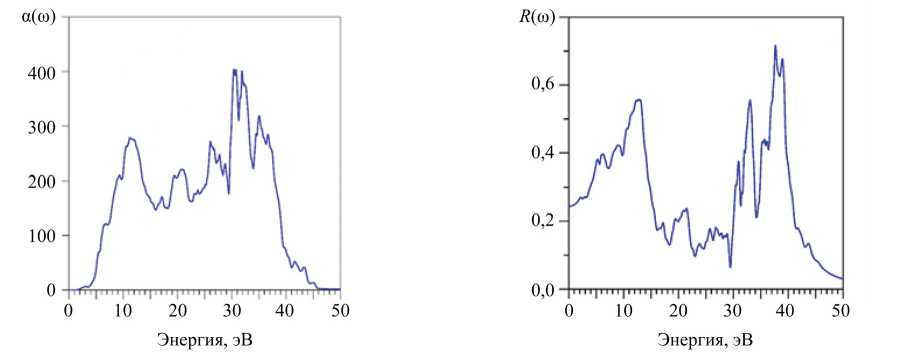
а) б)
Рис. 10. Рассчитанные в Eu2Zr2O7 коэффициенты: а — поглощения α(ω); б — отражения R (ω)
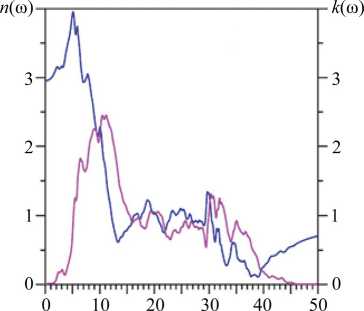
Энергия, эВ и(а) — ^(ш) а)
Ди)
ю
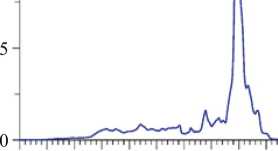
О 10 20 30 40 50
Энергия, эВ
-Д»)
б)
Рис. 11. Рассчитанные в Eu2Zr2O7: а — показатель преломления n (ω) и коэффициент экстинкции k (ω); б — спектр энергетических потерь электронов L( ω)
Обсуждение. Во всех исследованных в данной работе группах соединений основной вклад в верхнюю часть валентной полосы дают p-состояния атомов с наибольшей электроотрицательностью (халькогены, галогены), что можно объяснить уменьшением энергии связи p-состояний халькогенов и галогенов в кристалле, по сравнению с их связью в изолированном атоме, за счет перетекания к ним электронной плотности от атомов металлов из ближайшего окружения и вызванного этим увеличения экранировки этих состояний от ядра. S-состояния этих атомов образуют дно валентной полосы.
Для соединений с атомами, содержащими f-электроны, состояния 5s-симметрии, обладающие разными спинами, расщепляются по энергии под воздействием внутреннего магнитного поля 4f-электронов.
Прикладной аспект настоящей работы связан с тем, что применение новых материалов в квантовой электронике, оптоэлектронике, нелинейной оптике определяется откликом исследуемого кристалла на воздействие электронной волны с частотой, лежащей в оптическом или в ближнем и среднем ИК-диапазоне, то есть вблизи запрещенной щели полупроводника (E g ). Отклик кристалла можно описать с помощью тензора диэлектрической проницаемости (εi j ), расчет которого был проведен в настоящей работе для ряда сложных по составу трех- и четырехкомпонентных халькогенидов, галогенидов и оксидов. Академический интерес представляет влияние электронных состояний редкоземельного элемента, входящего в исследуемые пирохлоры, на электронно-энергетическое строение этих соединений.
Заключение. Достигнута основная цель работы — обобщены результаты исследования электронноэнергетической структуры различных групп соединений: халькогенидов Tl 3 TaS 4 , Tl 3 PS 4 , Sn 2 P 2 S 6 , InPS 4 , Cu 2 CdGeS 4 , Ag 2 CdSnS 4 , Ag 2 HgSnS 4 , галогенидов Cs 2 HgX 4 (X — Cl, Br, I) и APb 2 Br 5 (A — K, Rb), оксидов La 2 Zr 2 O 7 и Nd 2 Zr 2 O 7 и Ln 2 Zr 2 O 7 (Ln = La, Nd, Sm, Eu, Gd). Решены задачи определения влияния локальных парциальных электронных состояний на особенности электронной структуры исследованных соединений и их оптические свойства.
Подобные исследования важны в задачах моделирования новых материалов с заданными свойствами, так как позволяют определять, какие факторы оказывают основное влияние на возникновение таких свойств.
