Особенности применения метода масс-спектрометрии с индуктивно-связанной плазмой для анализа высокочистых твердых прекурсоров на основе пентаоксида ниобия

Автор: Елизарова Ирина Рудольфовна, Маслобоева Софья Михайловна
Журнал: Вестник Мурманского государственного технического университета @vestnik-mstu
Статья в выпуске: 3 т.16, 2013 года.
Бесплатный доступ
В работе приведены условия и особенности масс-спектрометрического с индуктивно-связанной плазмой анализа высокочистого и легированного гадолинием и эрбием пентаоксида ниобия. Рассчитаны метрологические параметры анализа: неопределенность, прецизионность, точность, пределы обнаружения аналитов. Установлено, что увеличение концентрации ниобия в пробах приводит к снижению интенсивности регистрируемого ионного тока аналитов. Показано наличие матричного эффекта от присутствия ниобия в пробе, который начинается с концентрации ионов ниобия, превышающей значение 14-15 ppm. Изотопы, не испытывающие влияния интерференции, имеют хорошую стабильность аналитического сигнала как в пробах с концентрацией менее 15 ppm, так и в пробах с более высокой концентрацией, при этом наблюдается дрейф интенсивности регистрируемого ионного тока во времени.
Пентаоксид ниобия высокочистый и легированный, масс-спектрометрический анализ с индуктивно-связанной плазмой, дрейф интенсивности, стандартные образцы, метрологические параметры
Короткий адрес: https://sciup.org/14294613
IDR: 14294613
Текст научной статьи Особенности применения метода масс-спектрометрии с индуктивно-связанной плазмой для анализа высокочистых твердых прекурсоров на основе пентаоксида ниобия
Развитие методов анализа неорганических соединений имеет решающее значение при создании конкурентоспособной технологии особо чистых веществ, используемых, например, в полупроводниковой, оптоволоконной технике, в микроэлектронике. В настоящее время возросли требования к способам аналитического контроля состава веществ как основного критерия их качества и пригодности. Если раньше в чистых веществах достаточно было определять содержание примесей на уровне 10-3-10-4 %, то теперь при производстве полупроводниковых материалов, веществ для получения оптических волокон и элементов микроэлектроники необходимо определять примеси с нижними границами содержания на уровне 10-5-10-7 %.
Оксиды ниобия находят широкое применение в различных областях техники. Так, высокочистый Nb 2 O 5 является основным компонентом в синтезе шихты ниобата лития (НЛ), используемой для выращивания монокристаллов, которые характеризуются уникальными электрофизическими, оптическими, нелинейно-оптическими и другими свойствами ( Кузьминов , 1975; 1987). Качество этих материалов неразрывно связано с контролем за содержанием примесей, определяемых в исходных продуктах на всех стадиях технологического получения монокристаллов НЛ.
Для анализа ниобиевых матриц используют радиохимические ( Шманенкова и др. , 1973; Caletka et al. , 1988), масс-спектрометрические ( Anderson et al. , 1992), а также атомно-спектрометрические методы с предварительным концентрированием примесей ( Anderson et al. , 1992; Stummeyer et al. , 1991). Как правило, концентрирование проводят экстракционными методами из фторидных сред ( Николаев,
Майоров , 1995; Das, Lahiri , 1991), нашли применение и сорбционные методы. Так, для отделения примесей используют анионообменник AG 1-8X ( Imakita et al. , 1990) и катионообменник AG 50W-8X ( Stummeyer et al. , 1991).
Масс-спектрометрический анализ с индуктивно-связанной плазмой (МС-ИСП) соединений ниобия, в том числе легированных, может быть сопряжен с серьезными трудностями ввиду существенного матричного влияния ниобия на интенсивность аналитического сигнала. Эффект снижения интенсивности может иметь как спектральную природу, так и не спектральную. При анализе наиболее существенным фактором, вызывающим дрейф интенсивности аналитического сигнала к занижению, может оказаться плазмохимическое образование оксидных соединений ниобия и их осаждение в виде стекловидного слоя вокруг отверстий скиммера и сэмплера. Первостепенным в этом случае становится выбор предельной солевой концентрации раствора, подаваемого в систему ввода пробы масс-спектрометра. Возможны и другие причины, например, удаление изотопов легких аналитов из зоны ионной оптики и сепарирования вместе с матричными элементами из ионного потока, имеющими значительные массы. Обзор ( Карандашев и др ., 2012) приводит упоминание многих работ, где исследовались матричные эффекты и способы их нивелирования.
Снижение аналитических сигналов при анализе примесей в пентооксиде ниобия показано в работе ( Елизарова, Рыжухина , 2010). Обнаружен матричный эффект при анализе Ca, Fe, Ti, Mg, Si в растворах с содержанием ниобия выше 15 ppm. Наибольшее падение интенсивности при наличии в пробе ионов ниобия наблюдали, начиная с соотношения элемент : ниобий = 1 : 50. Для этих же элементов наблюдался существенный дрейф интенсивности – уменьшение на 30-50 % в течение часа непрерывного пропускания пробы в системе ввода образца масс-спектрометра.
В работе ( Grebneva et al. , 1998) методом атомно-эмиссионной спектрометрии с индуктивносвязанной плазмой (АЭС-ИСП) определяли примеси в высокочистом ниобии после сорбционного отделения матрицы пробы на сорбенте Полиоргс VII. Авторам работы удалось провести анализ с пределами обнаружения от 0.1 до 10 ppm.
МС-ИСП анализ после электротермического испарения порошков пентаоксидов ниобия в графитовой кювете с добавками фторирующего агента представлен в работе ( Shengging et al. , 2004). Здесь же дано описание возможных полиатомных интерференций. Авторы указывают на наличие матричного эффекта при концентрации Nb в растворе пробы, начиная с 140 ppm. При определении Ti, Ta, W, Cr, Ni, Cu, Mn предел обнаружения составил 0.026-1.1 ppb.
Таким образом, практически все литературные данные, имеющиеся на сегодняшний момент, приводят сведения об определении концентрации примесей не на фоне высокого содержания в пробе ниобия, а при использовании его предварительного отделения. Это также отмечено в работах ( Kozono, Haraguchi , 2007; Kong-Quan et al. , 2004). Авторы указывают, что пределы обнаружения большинства примесных элементов после процедур разделения составляют величины на уровне ppb-ppm.
Данная работа посвящена изучению особенностей применения метода масс-спектрометрии с индуктивно-связанной плазмой для определения Ta, Pb, W, Mn, Ni, Cr, Co, V, Mo, Cu, Ti, Si, Al, Mg, Fe, Zr, Sn, Ca, Gd, Er в твердых прекурсорах на основе чистого и легированного различными примесями пентаоксида ниобия и нахождению оптимальных условий анализа этих продуктов.
-
2. Методика и результаты исследования
В работе использовали пентаоксиды ниобия, полученные при экстракционной переработке редкометалльного сырья в ИХТРЭМС КНЦ РАН, а также сертифицированный Nb 2 O 5 Соликамского магниевого завода, ГСО 7357-97. Вскрытие оксидов ниобия осуществляли кислотным разложением с использованием дистиллированных HF и HNO 3 при объемном соотношении 1 : 3. Навеску массой около 70 мг помещали в полипропиленовую пробирку, добавляли сначала фторводородную, а затем азотную кислоту, закрывали пробкой на резьбовом соединении и выдерживали на водяной бане при температуре 70-90 °С до полного растворения. Nb(OH) 5 вскрывали в НNO 3 при таких же условиях. Дистиллированные кислоты получали перегонкой ниже температуры кипения с использованием системы BERGHOF. Для приготовления растворов применяли деионизованную воду (Milli-Q A10 Water Systems) пяти ступеней очистки. Все разбавления проводили 2 %-й азотной кислотой.
Пентаоксиды ниобия, легированные гадолинием и эрбием, вскрыть в полипропиленовых пробирках на водяной бане не удалось. Значительная часть пробы оставалась в виде нерастворимого осадка, что было вызвано упрочнением решетки оксида после легирования и прокаливания при Т > 1273 К. Полностью перевести навеску пробы массой ≈ 100 мг в раствор удалось при использовании в качестве вскрывающего реагента смеси 15 мл серной кислоты и 5 г сульфата аммония при нагревании в стеклянном стакане, накрытом часовым стеклом. Длительность вскрытия – до 8 часов. Раствор пробы переводили деионизованной водой в полипропиленовую пробирку, добавляли 1-2 мл пероксида водорода медицинского и доводили до метки 50 мл 2 %-й азотной кислотой.
Анализ проб проводили в условиях "чистого блока" на квадрупольном масс-спектрометре ELAN 9000 DRC-e. (Perkin Elmer, США). Настройка и оптимизация режимов работы прибора производилась при помощи программного обеспечения ЕLAN с использованием раствора на основе 2 %-й HNO 3 , содержащего 10 ppb: Be, Ce, Co, Bi, Ni, Pb, In, Mg, U (Standard solution, Perkin Elmer, США). Для построения градуировочных зависимостей применяли стандартные мультиэлементные растворы компании Perkin Elmer ("Multi-element ICP-MS Calibration Std"). Фоновую концентрацию ниобия в растворах градуировки задавали добавками раствора, полученного в результате параллельного пробам кислотного вскрытия Nb 2 O 5 марки "А" Соликамского магниевого завода. Контролируемые примеси: Fe, Сa, Si, Ta, Pb, Mg, Ti, Nb, W, V, Mo, Co, Ni, Na, Al, Sb, Mn, Sn, Cr, Cu, Gd, Pr, Nd, Tb, Dy, Er.
Градуировочные зависимости строили из начала координат интенсивность сигнала – концентрация, коэффициент аппроксимации полученных линейных зависимостей составлял 0.99980.9999. Среднеквадратичное отклонение результатов анализа в параллельных измерениях составляло менее 3 %.
При приготовлении растворов для построения градуировочных характеристик объем исходных стандартных растворов Perkin Elmer "Multi-element ICP-MS Calibration Std" № 2, 3, 5, указанный в табл. 1, отбирали в пластиковые пробирки с добавленной в них предварительно фоновой концентрацией ионов ниобия, доводили до метки 2 %-й деионизованной азотной кислотой и тщательно перемешивали. 2 %-й раствор азотной кислоты с добавкой фонового матричного компонента – ниобия – использовали как холостой раствор.
Проверку результатов анализа проводили методом добавок и по внешним стандартам, в качестве которых использовали растворы на основе ГСО состава ионов контролируемых аналитов, ГСО пентаоксида ниобия 7357-97 и в качестве образца сравнения применяли сертифицированный пентаоксид ниобия (Соликамский магниевый завод, Пермская область).
Выбор контролируемых примесей в чистом виде и легированных различными элементами петаоксидах ниобия определялся в соответствии с требованиями ТУ на всех этапах получения экстракционным методом Nb 2 O 5 .
Условия анализа: аргоновая плазма мощностью 1300-1350 Вт, поток газа распылителя 0.90-1.2 л × мин-1, напряжение ионной линзы 6-11 В, содержание оксидных и двухзарядных ионов не более 3 %, уровень интенсивностей по In, U, Mg, Ce в соответствии с паспортными данными.
Таблица 1. Растворы для построения градуировочных характеристик при определении элементов методом МС-ИСП
|
Объем аликвоты, 10-6 × дм3 |
Массовая доля аналитов в стандартном растворе, ppm (мг × дм-3) |
Объем градуировочного раствора, см3 |
Массовая доля аналитов в градуировочном растворе, ppm (мг × дм-3) |
|
40 |
10 |
10 |
0.04 |
|
60 |
10 |
10 |
0.06 |
|
80 |
10 |
10 |
0.08 |
|
100 |
10 |
10 |
0.1 |
|
200 |
10 |
10 |
0.2 |
|
300 |
10 |
10 |
0.3 |
|
400 |
10 |
10 |
0.4 |
|
500 |
10 |
10 |
0.5 |
|
600 |
10 |
10 |
0.6 |
Характер дрейфа интенсивности сигналов определяли, используя растворы аналитов (0.2 ppm) в течение 1 часа непрерывных измерений пробы, содержащей 56 ppm Nb. Полученные данные представлены на рис. 1.
При анализе ниобийсодержащих проб важным фактором, влияющим на величину погрешности при определении, являлось осаждение ниобия вблизи отверстий семплера и скиммера. Образующиеся оксиды остекловывались, что приводило к уменьшению диаметра отверстий и влекло за собой снижение чувствительности прибора из-за уменьшения интенсивности регистрируемого ионного тока.
Следующей, не менее важной причиной являлось наличие матричного эффекта, создаваемого ионами ниобия по отношению к легким элементам. Они оказывались "запертыми" между ионизированными изотопами ниобия и отбрасывались вместе с ними квадрупольной системой, что приводило к погрешностям определения, занижая результат.
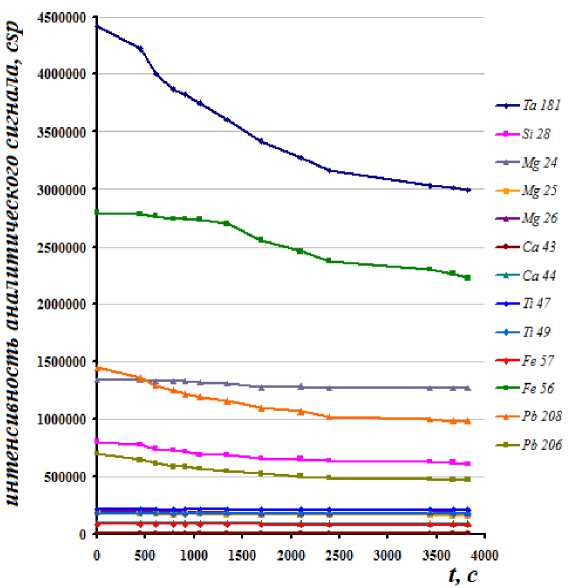
Рис. 1. Дрейф интенсивности сигналов аналитов (0.2 ppm) в течение 1 часа непрерывных измерений пробы, содержащей 56 ppm Nb
Проверка правильности анализа по стандартному образцу состава ионов железа (ГСО 8032-94) показала, что определения концентрации железа на массе изотопов 54Fe и 56Fe дают завышение результатов соответственно в 3.6 и 2.0 раза, что вызвано интерференционным наложением массы полиатомных ионов 40Ar14N и 40Ar16O. Интересно было заметить, что кроме такого интерференционного искажения интенсивности анализируемого сигнала для ионов 54Fe и 56Fe наблюдался и их значительный дрейф (рис. 1), подобная картина наблюдалась и для других аналитов, исключенных из списка изотопов (рис. 2). Для 57Fe такого дрейфа не было, поэтому именно он был использован для определений концентраций железа. Наложение изотопа 28SiH на 29Si дискриминировало сигнал последнего, кроме того, его природная распространенность мала (4.67 %), поэтому определение кремния проводили на 28Si. При анализе свинца и тантала учитывали значительный дрейф их интенсивности в течение времени проведения измерений, вызванный не столько наличием ниобия в матрице пробы, сколько собственным поведением иона. Как и ниобий, эти ионы легко осаждаются на рабочих поверхностях в зоне интерфейса. Причина дрейфа не выяснялась, его влияние устраняли периодической переградуировкой, как только отклонения начинали превышать 10 %. Дрейф интенсивности для тантала не превышал 6 % в течение первых 8 минут непрерывного пропускания пробы через систему ввода. При отклонениях на 3-10 % от значения, установленного в растворе сравнения (раствор стандартного образца состава ионов свинца и тантала ГСО 7012-93 и ICP-AES and ICP-MS Standard, Ta (Perkin Elmer)), проводили математическую коррекцию результатов. Кроме того, для контроля приборного дрейфа интенсивностей в пробы вводили известную концентрацию In в качестве внутреннего стандарта.
Установлено, что матричный эффект от присутствия ниобия в пробе начинается с концентрации, превышающей значение 14-15 ppm, до этого значения изменение концентрации аналитов находится в пределах погрешности метода (рис. 2). Зависимости, приведенные на рис. 2, получали при промывке системы ввода пробы после каждого измерения холостым раствором в течение 2-3 минут, чтобы исключить изменение аналитического сигнала за счет дрейфов. При этом, изотопы, не испытывающие влияние интерференции, имеют хорошую стабильность аналитического сигнала как в пробах с концентрацией менее 15 ppm, так и в пробах с более высокой концентрацией.
Для определения концентрации примесей были выбраны 93Nb, 181Ta, 47Ti, 49Ti, 183W, 184W, 57Fe, 44Ca, 26Mg, 51V, 27Al, 28Si, 55Mn, 95Mo, 97Mo, 65Cu,59Co, 61Ni, 62Ni, 121Sb, 118Sn, 119Sn, 206Pb, 207Pb, 23Na, 29K, 52Cr, 53Cr, 66Zn, 68Zn, 91Zn.
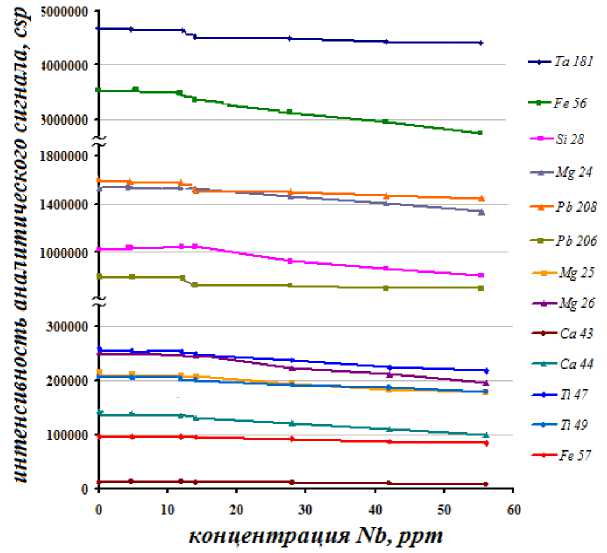
Риc. 2. Падение интенсивности аналитического сигнала контролируемых элементов (по 0.2 ppm) при изменении концентрации Nb в пробе
При анализе свинца учитывали значительный дрейф его интенсивности в течение времени проведения измерений, вызванный не наличием ниобия в матрице пробы, а собственным поведением иона. Причина дрейфа не выяснялась, его влияние устраняли периодической переградуировкой, как только отклонения начинали превышать 10 %. При отклонениях на 3-10 % от значения, установленного в растворе сравнения (раствор стандарта ГСО 7012-93), проводили математическую коррекцию результатов. Кроме того, для учета приборного дрейфа интенсивностей в пробы вводили известную концентрацию In в качестве внутреннего стандарта.
Была проведена проверка сходимости результатов определения при построении градуировочных графиков на фоне растворов, содержащих ниобий и при его отсутствии. Наиболее точные результаты анализа растворов с концентрацией ионов ниобия до 70 г × дм-3 получены при использовании градуировочных зависимостей, построенных при добавлении стандартного раствора Perkin Elmer (0.1-0.6 ppm контролируемых элементов) к фоновому раствору, содержащему до 30-56 ppm Nb.
Для оценки неопределенности анализа в условиях промежуточной прецизионности (изменяющиеся факторы – время и оператор) были проанализированы стандартные образцы состава оксида ниобия ГСО 7357-97; ОСО 95.333-91. Результаты определений представлены в табл. 2, 3.
Таблица 2. Результаты масс-спектрометрического анализа государственного стандартного образца СO-3 (ГСО 7357-97)
|
Элемент |
Аттестованное значение, % |
Получено МС-ИСП, % |
Неопределенность* δ , % |
Относительное стандартное отклонение**, % |
|
W |
(4.8±1.5) × 10-3 |
3.6 × 10-3 |
2.9 |
1.1 |
|
Fe |
(0.99±0.12) × 10-3 |
0.97 × 10-3 |
2.1 |
3.8 |
|
Al |
(0.99±0.25) × 10-3 |
0.75 × 10-3 |
1.3 |
1.8 |
|
Ca |
(0.99±0.11) × 10-3 |
0.98 × 10-3 |
1.0 |
1.1 |
|
V |
(4.9±0.6) × 10-4 |
4.8 × 10-4 |
1.0 |
2.1 |
|
Co |
(4.9±1.2) × 10-4 |
4.7 × 10-4 |
4.1 |
3.3 |
|
Mn |
(4.9±0.8) × 10-4 |
4.3 × 10-4 |
4.8 |
1.2 |
|
Cu |
(4.9±0.23) × 10-4 |
4.7 × 10-4 |
4.1 |
1.3 |
|
Ni |
(4.9±1.0) × 10-4 |
4.8 × 10-4 |
2.0 |
0.5 |
|
Элемент |
Аттестованное значение, % |
Получено МС-ИСП, % |
Неопределенность* δ , % |
Относительное стандартное отклонение**, % |
|
Cr |
(4.9±0.6) × 10-4 |
4.8 × 10-4 |
2.0 |
4.4 |
|
Ti |
(2.2±0.3) × 10-4 |
2.5 × 10-4 |
1.0 |
1.2 |
|
Mg |
(4.9±1.3) × 10-4 |
4.2 × 10-4 |
2.3 |
3.1 |
|
Pb |
(5.0±1.2) × 10-4 |
0.6 × 10-3 |
1.0 |
1.8 |
|
Zr |
(4.94±0.05) × 10-4 |
4.87 × 10-4 |
0.5 |
3.8 |
|
Sn |
(0.99±0.17) × 10-4 |
0.85 × 10-4 |
3.7 |
0.7 |
|
Zn |
(0.99±0.22) × 10-3 |
0.80 × 10-3 |
3.8 |
1.8 |
|
Bi |
(4.9±0.4) × 10-4 |
4.7 × 10-4 |
4.1 |
1.7 |
|
Mo |
(5.0±1.2) × 10-5 |
4.4 × 10-5 |
2.3 |
1.8 |
|
As |
(3.0±1.0) × 10-3 |
1.9 × 10-3 |
5.0 |
0.9 |
|
Ta |
0.12 |
0.8 |
Анализ государственных стандартных образцов ГСО 7357-97; ОСО 95.333-91 (табл. 2, 3) показал хорошую сходимость результатов измерений с аттестованными значениями концентраций. Это оценивали по величине неопределенности анализа δ (%). Она не превысила 5 % для ГСО 7357-97. Для ОСО 95.333-91 погрешность измерения не более 5 %, кроме железа.
Пределы обнаружения для контролируемых примесей составили 10-50 ppt, их определяли по 3σ-критерию, используя в качестве фоновых интенсивностей аналитического сигнала аналитический отклик раствора кислот вскрытия проб.
Повторяемость результатов измерений методом МС-ИСП оценивали по данным относительного стандартного отклонения, которое рассчитывали при каждом измерении с использованием возможностей программного обеспечения масс-спектрометра. Статистика проводилась по данным трех реплик в режиме анализа "скачок по пикам" (3 измерения на каждом пике), сканирования пиков проводилось трижды в каждой реплике. Таким образом, при детектировании один результат измерения получали по данным 27 регистраций аналитического сигнала. При этом при анализе каждой разбавленной пробы проводили три единичных измерения. Пробы вскрывали в трех параллелях, в каждой из которых делали три разбавления.
Таблица 3. Результаты масс-спектрометрического анализа государственного стандартного образца CТ (ОСО 95.333-91)
|
Элемент |
Аттестованное значение, % |
Получено МС-ИСП, % |
Неопределенность* δ , % |
Относительное стандартное отклонение**, % |
|
W |
0.022 |
2.1 × 10-2 |
4.5 |
2.4 |
|
Fe |
0.101 |
0.108 |
6.9 |
4.8 |
|
Al |
0.015 |
1.6 × 10-2 |
3.0 |
4.9 |
|
Ca |
0.184 |
0.186 |
1.1 |
4.9 |
|
Co |
2.9 × 10-5 |
4.1 |
||
|
Mn |
5.6 × 10-4 |
5.1 |
||
|
Cu |
6.8 × 10-4 |
4.8 |
||
|
Ni |
4.9 × 10-3 |
5.2 |
||
|
Cr |
3.9 × 10-3 |
4.2 |
||
|
Ti |
0.285 |
0.290 |
1.7 |
3.0 |
|
Mg |
1.1 × 10-2 |
3.9 |
||
|
Pb |
2.7 × 10-4 |
4.2 |
||
|
Zr |
1.5 × 10-4 |
4.9 |
||
|
Zn |
1.2 × 10-3 |
4.5 |
||
|
Bi |
3.5 × 10-6 |
5.4 |
||
|
Mo |
0.0025 |
2.6 × 10-3 |
4.0 |
3.3 |
|
As |
1.3 × 10-3 |
4.9 |
||
|
Ta |
4.9 × 10-2 |
1.5 |
