Особенности включения стабильного изотопа 18О в сильнокислой среде в карбоксильные группы пептидов

Автор: Манойлов А.В., Новиков А.В., Бубляев Р.А., Серебрякова М.В., Козьмин Ю.П., Краснов Н.В., Миргородская О. а
Журнал: Научное приборостроение @nauchnoe-priborostroenie
Рубрика: Масс-спектрометрия для биотехнологии. Интерпретация данных, методология, применение
Статья в выпуске: 4 т.18, 2008 года.
Бесплатный доступ
С использованием ESI-MS и MALDI-MS изучен процесс изотопного обмена 16О на 18О в карбоксильных группах пептидов. Показано, что лимитирующей стадией при обмене является протонизация карбоксильной группы. Причем протонизация зависит от кислотности карбоксильных групп и кислотности среды. Определены оптимальные условия для получения 18О-содержащих пептидов и производных аминокислот, которые могут быть использованы в качестве стандартов для количественного анализа. Оценена стабильность вводимой метки в условиях, близких к физиологическим, а также в средах, наиболее часто используемых при работе с образцами в экспериментах с применением ВЭЖХ.
Короткий адрес: https://sciup.org/14264575
IDR: 14264575 | УДК: [577.1:
Features of the stable 18O isotope inclusion into peptide carboxyl groups in highly acidic media
Using ESI-MS and MALDI-MS the process of isotope exchange of 16О for 18О in peptide carboxyl groups was studied. It was shown, that carboxyl group protonization is a limiting stage in this exchange. Protonization depends on carboxyl group acidity as well as acidity of the medium. Optimum conditions for the production of 18O-containing peptides and amino acid derivatives which can be used as standards for the quantitative analysis are determined. Stability of the introduced label in the conditions approaching physiological, and in media commonly used in experimental work with specimen using HPLC and ESI-MS was evaluated.
Текст научной статьи Особенности включения стабильного изотопа 18О в сильнокислой среде в карбоксильные группы пептидов
Количественное определение отдельных компонентов протеом в биомедицинских исследованиях является актуальной задачей [1–2]. Это связано с тем, что мониторинг изменения количественных характеристик отдельных компонентов протеом позволит не только расширить понимание патологических процессов, но и может использоваться в диагностических целях.
В настоящее время все больше для количественных измерений при протеомном анализе используются масс-спектрометрические методы, такие как MALDI-MS и ESI-MS [3–5]. Это прежде всего связано с тем, что по достоверности идентификации компонентов протеом и универсальности масс-спектрометрии нет равных. Однако в области масс-спектрометрического количественного анализа имеются сложности, связанные с тем, что интенсивности сигналов не соответствуют даже относительным концентрациям, а сами интенсивности не воспроизводятся. При этом интенсивности изотопных распределений хорошо воспроизводятся и, более того, соответствуют теоретически рассчитанным отношениям. Это используется для количественных измерений посредством внесения в качестве внутреннего стандарта соединения, химически идентичного измеряемому, но с отличающимся изотопным составом [5–7]. Сопоставляя интенсивности изотопного распределения смеси анализируемого вещества и стандарта можно вычислить их количественное соотношение.
Среди известных способов получения стандартов наибольший интерес вызывает метод прямого ввода изотопов 18О в карбоксильные группы пеп- тидов. Суть метода состоит в обмене 16О на 18О в кислой среде в присутствии Н218О [8]. К преимуществам данного подхода можно отнести его универсальность. Кроме того, данный подход позволяет полностью сохранить химическую структуру соединения после введения изотопа 18О.
Для понимания механизма процесса включения изотопа 18О и для практического использования этого подхода для получения и использования 18О-содержащих продуктов в качестве стандартов в настоящей работе проведен ряд экспериментов на модельных пептидах:
-
• определены относительные скорости обмена изотопа 16О на 18О в различных типах карбоксильных групп;
-
• определены зависимости скорости обмена от характеристик среды;
-
• сопоставлены скорости обмена со скоростями возможных побочных процессов, связанных с разрывом отдельных пептидных связей и дезамидированием.
Эксперименты проводились с использованием двух типов масс-спектрометрии — MALDI-MS и ESI-MS.
РЕЗУЛЬТАТЫ ЭКСПЕРИМЕНТА И ИХ ОБСУЖДЕНИЕ
Для экспериментов в работе использованы производные аминокислот и пептидов, представленные в табл. 1. Прежде всего на примере даларгина оценена скорость обмена в зависимости от концентрации ТФУ и температуры. Контроль за обменом осуществлялся с использованием ESI-MS.
Табл. 1. Характеристика пептидов, использованных для обмена 16О на 18О
|
№ п/п |
Пептид |
Аминокислотная последовательность |
М, Да |
|
1 |
TA |
N-(Tos)R |
328.9 |
|
2 |
Даларгин |
YAGFLR |
725.39 |
|
3 |
Br |
RPPGFSPFR |
1059.56 |
|
4 |
Ang II |
DRVYIHPF |
1046.5 |
|
5 |
Ang I |
DRVYIHPFHL |
1295.68 |
|
6 |
P1 |
AcDAEFR |
678.30 |
|
7 |
P2 |
HDSGYEVHHQK |
1334.61 |
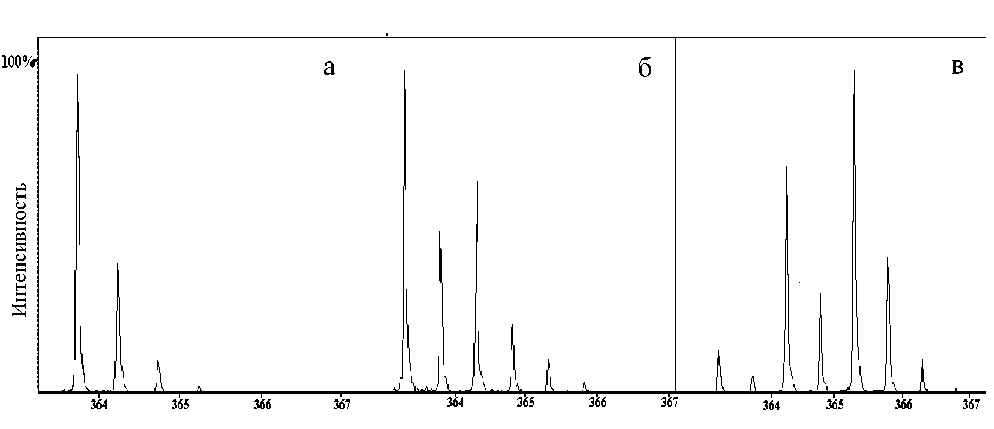
Рис. 1. Масс-спектры даларгина: а — до обмена; б — после обмена H 2 18O при 50 оС в течение 30 мин; в — после обмена при 70 оС в течение 180 мин
С помощью масс-спектрометрии также контролировалось наличие возможных продуктов гидролиза. На рис. 1 в качестве примера представлены масс-спектры даларгина до обмена (а) и после обмена при 50 оС в течение 30 минут (б), и при 70 оС в течение 180 минут (в). Обмен проводили в смеси 50 мкл H 2 18O и 3 мкл 100 %-й ТФУ.
Для данного пептида, в котором присутствует одна карбоксильная группа, максимально возможно включение двух атомов 18О. Из соотношения интенсивностей ионов даларгина до и после обмена можно оценить степень включения метки на разных глубинах обмена. Для этого ввели величину n18O. С помощью специального программного обеспечения были определены степени включения метки в даларгин при условиях рис. 1: спектр а — n18O = 0; спектр б — n18O = 0.34; спектр в — n18O = = 1.44. Для получения зависимостей скорости изотопного обмена как от концентрации кислоты, так и температуры были рассчитаны в каждом случае степени включения метки (см. рис. 2 и 3).
Из полученных данных можно для пептидов с С-концевыми карбоксильными группами выбрать подходящие условия получения стандартов с достаточной степенью обмена для количественного анализа.
Использование ТФУ в качестве катализатора для изотопного обмена обусловлено главным образом тем, что она имеет высокую степень диссоциации. Кроме того, ТФУ обладает высокой летучестью,
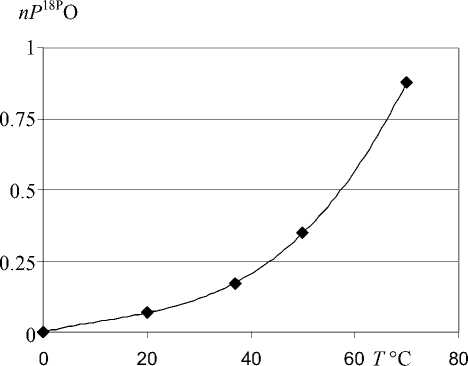
Рис. 2. Зависимость степени включения P18PO в даларгин за 30 мин в 5.7 %Bоб.B ТФУ от температуры
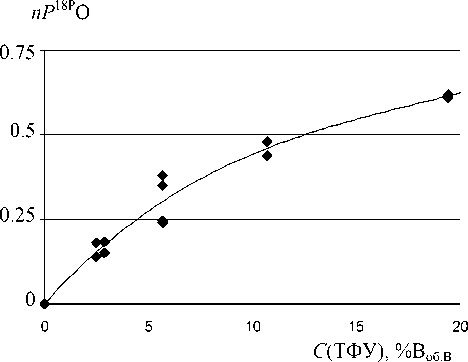
Рис. 3. Зависимость степени включения P18PO в даларгин от концентрации ТФУ при температуре 50P оP что позволяет легко удалить избыток, например, на вакуумной центрифуге. Полученные стандарты после высушивания могут храниться в условиях, аналогичных хранению других пептидов. Перед использованием для количественного анализа образцы растворяются в обычной воде и могут храниться достаточно долго. Однако перед их использованием необходимо каждый раз определять соотношение изотопов в стандарте, а программное обеспечение учтет любые изменения в соотношениях.
В специальных экспериментах была оценена стабильность метки в средах, наиболее часто ис- пользуемых при работе с образцами в биохимических и других экспериментах, например при ВЭЖХ. Результаты этих экспериментов представлены на рис. 4 в виде диаграмм.
Из полученных данных следует, что введенная изотопная метка достаточно стабильна в широком диапазоне рН, в том числе при типичных условиях ВЭЖХ, используемой для разделения пептидов, и, что особенно важно для ESI-MS, при обессоливании образцов.
Как правило, практически для получения нужных стандартов для пептидов нами использовалась концентрация ТФУ 5.7 % об. и Т = 50 оС, время для разных образцов выбиралось экспериментально по результатам масс-спектрометрического анализа. В связи с тем, что в этих условиях, хотя и сохранялись пептидные связи, но можно ожидать частичное дезамидирование, нами проведена оценка скоростей этого процесса на примере дезамидирования данзильного производного глутамина DnsGln и превращения его в кислоту DnsGlu. За дезамидированием следили с помощью ESI-MS по появлению продуктов, моноизотопная масса которых на 1 Да больше, чем у исходного соединения. При оценке учитывали природное изотопное распределение как исходного соединения, так и его продукта дезамидирования. Специальным экспериментом для DnsGln и DnsGlu определена пропорциональность концентраций и интенсивностей соответствующих моноизотопных ионов. С учетом всех этих факторов оценка скорости дезамидирования в выбранных условиях представлена на рис. 5.
И как следует из этого эксперимента, при выбранных нами условиях дезамидирование достаточно заметно, и его необходимо учитывать. Для нивелирования этого процесса можно выдерживать анализируемый образец в аналогичных условиях. Вместе с тем наличие дезамидированной формы может быть легко учтено с помощью программного обеспечения, тем более что дезамидирование в этом случае будет сопровождаться дополнительным включением изотопа 18О в образец.
Отметим, что во всех приведенных выше экспериментах обмен осуществлялся исключительно в С-концевых карбоксильных группах. Вместе с тем в пептидных фрагментах широко представлены карбоксилсодержащие аминокислоты — Asp и Glu. Наличие этих аминокислотных остатков будет способствовать дополнительному включению изотопа 18О. В каждой из присутствующих в пептидах карбоксильной группе осуществляется обмен изотопа 16О на 18О, что способствует увеличению суммарного количества изотопа18О — в максимуме по 2 на каждую карбоксильную группу. Примеры с результатами такого обмена ряда пептидов (см. табл. 1) в сопоставимых условиях представлены на рис. 6. В этих экспериментах тип
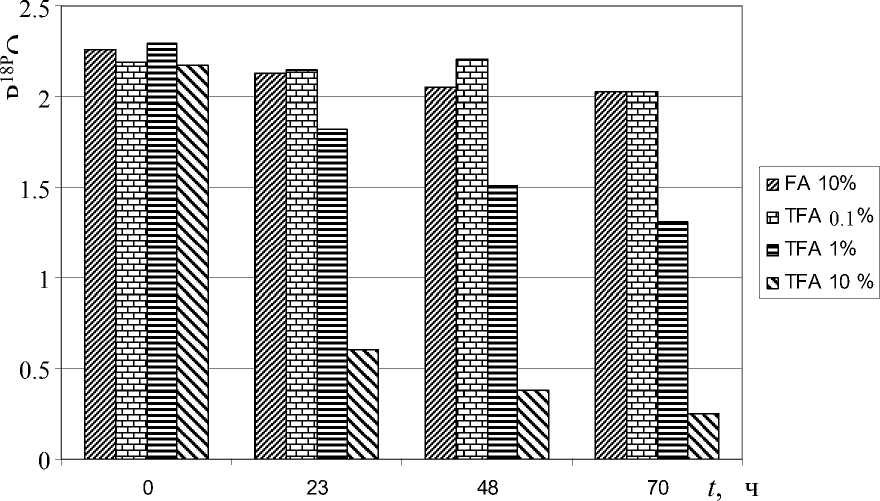
Рис. 4. Зависимость степени включения метки 18O в различных средах от времени.
FA — муравьиная кислота, TFA — трифторуксусная кислота, T = 25 оС
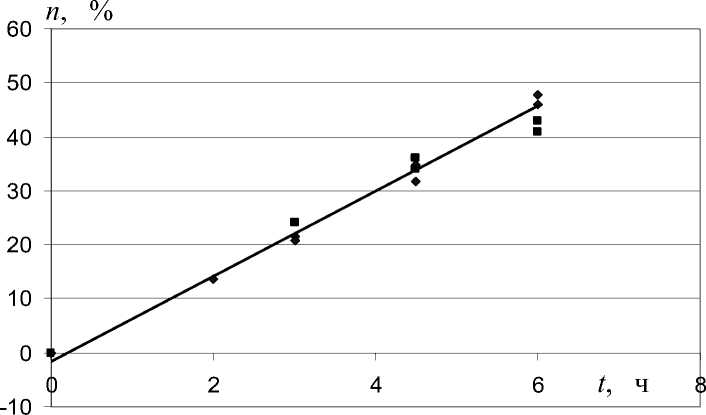
Рис. 5. Зависимость степени превращения данзилглута-мина(амида) в кислоту при T = 25 PоPС от времени.
♦ — МС; ■ — ВЭЖХ
пептидов, и температура, и время обмена выбирались таким образом, чтобы можно было сравнить скорости обмена в разных типах карбоксильных групп в сопоставимых условиях.
В соответствии с данными, представленными в табл. 2 и на рис. 1П, 2П (Приложение), наибольшее суммарное включение изотопа и более широкое распределение 18О-изотопсодержащих типов пептидов, содержащих от одного до трех (18О 1 –
18О 3 ), наблюдается в пептидах с наибольшим количеством карбоксильных групп. Однако для пептида с одной карбоксильной группой — Br и для двух пептидов с 2 карбоксильными группами — Ang I и Ang II включения изотопа отличаются в меньшей степени.
Для выявления особенностей включения 18О в различные карбоксильные группы и оценки распределения изотопа при включении в пептиды,
Табл. 2. Относительное содержание изотопа 18O в пептидах и в их отдельных аминокислотах
|
Пептид |
Время, мин |
Содержание изотопа 18O в пептиде (%) |
Содержание изотопа 18О для аминокислоты (%) |
||||||
|
18O 0 |
18O 1 |
18O 2 |
18O 3 |
18 4 |
D |
E |
C-terminus |
||
|
Br |
50 |
78.6 |
21.4 |
— |
— |
— |
— |
— |
21.4 |
|
Ang II |
74.1 |
25.9 |
— |
— |
— |
11 |
— |
18 |
|
|
Ang I |
63.3 |
36.7 |
— |
— |
— |
13 |
— |
20 |
|
|
P1 |
35.62 |
43.53 |
18.29 |
2.56 |
— |
18 |
33 |
14 |
|
|
P2 |
39.86 |
39.54 |
15.56 |
5.04 |
— |
— |
— |
— |
|
|
Br |
100 |
50.93 |
43.71 |
6.82 |
— |
— |
— |
— |
49 |
|
Ang II |
47.08 |
39.34 |
11.56 |
— |
— |
23 |
— |
36 |
|
|
Ang I |
47.22 |
38.45 |
13.10 |
1.23 |
— |
18 |
— |
32 |
|
|
P1 |
5.96 |
27.34 |
38.33 |
22.99 |
5.37 |
— |
— |
— |
|
|
P2 |
11.10 |
33.29 |
38.18 |
13.20 |
4.23 |
— |
— |
— |
|
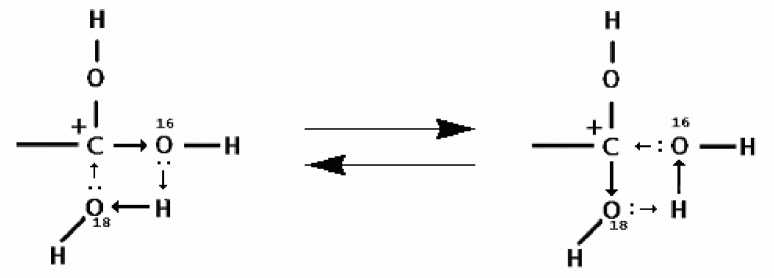
Рис. 7. Предполагаемая схема изотопного обмена
содержащие более одной карбоксильной группы, для трех пептидов — Ang I, Ang II и P1 — была проведена фрагментация с использованием MALDI-MS/MS. Масс-спектры фрагментации Ang I, Ang II и P1 после обмена для обоих времен (50 и 100 мин) представлены на рис. 3П–5П (в Приложении).
В каждом из спектров фрагментации были выбраны ионы, содержащие исключительно одну из карбоксильных групп, и в них по соотношению изотопных пиков оценивалась степень обмена.
Для оценки степени обмена из данных, представленных на рис. 3П и рис. 4П (см. Приложение), для Ang II и Ang I — пептидов, имеющих одинаковую последовательность с 1 по 8 амино- кислоту, был выбран одинаковый ион (b28) для N-концевых аминокислот с m/z = 354.1. Для С-концевых аминокислот выбраны соответственно y´´ ионы с m/z = 263.1 и m/z = 269.1. Рассчитанное из этих данных содержание изотопов в отдельных аминокислотах в пептидах Ang II и Ang I при каждом времени обмена представлено в табл. 2.
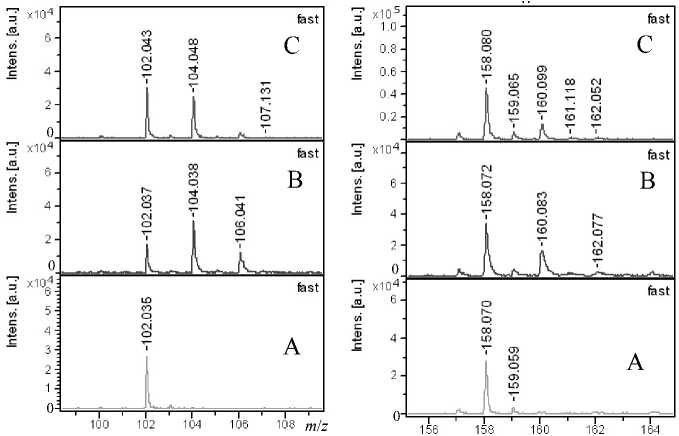
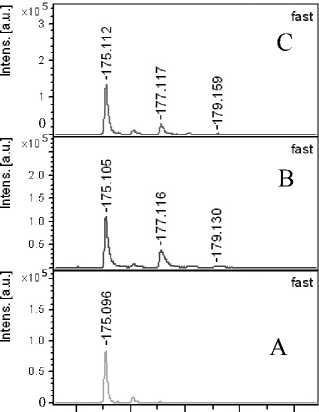
Для пептида Р1 выбраны в спектрах ионы, соответствующие отдельным аминокислотам Glu, AcAsp и Arg с m / z = 102.05, m / z = 158.05 и с m / z = = 175.05 соответственно (см. рис. 5П–6П в Приложении).
Рассчитанные для Р1 из интенсивностей фраг-ментных ионов соотношения 18О в аминокислотных остатках ионов Glu, AcAsp и Arg при разных глубинах обмена представлены в табл. 2.
Из полученных данных следует, что наибольшая скорость обмена наблюдается в карбоксильной группе Glu. Для Asp скорость обмена в Р1 несколько выше, чем в С-концевой группе. Аналогичная степень обмена наблюдается в пептиде Р2, в котором присутствуют все три типа карбоксильных групп. Причем в одном случае Asp, хотя и является N-концевой аминокислотой, по сути таковой не является, т. к. ее аминогруппа ацелирована. Это соответствует ее нахождению внутри пептидной цепи, как в пептиде Р2. Однако если Asp является N-концевой группой, то ее скорость обмена снижается в соответствии с ожидаемым увеличение рКа из-за наличия вблизи свободной аминогруппы.
Зависимость скорости обмена от рКа карбоксильной группы была дополнительно подтверждена экспериментально сравнением скоростей обмена у Ala (рКа 2.35) и GlyAla (рКа 3.7). По данным ESI-MS при сопоставимых условиях скорость обмена у GlyAla в 3 раза выше, чем у Ala. Из полученных данных следует, что скорость обмена в карбоксильной группе тем выше, чем слабее ее кислотность.
Совокупность полученных данных позволяет предложить следующий механизм обмена (рис. 7). Лимитирующей стадией при обмене является протонирование карбоксигруппы, которое ускоряется при ослаблении кислотности карбоксильных групп и повышении кислотности среды, т. к. в данном процессе карбоксильная группа вынуждена играть роль основания. При этом протонированная карбоксигруппа оказывается сольватирована водой, содержащей изотоп 18О, с образованием промежуточного комплекса. Этот комплекс не только стабилизирует существование образующегося катиона, но и способствует самому акту протонирования за счет электроотрицательных свойств кислорода воды.
Обмен в карбоксильной группе 16О на 18О происходит в результате циклического электронного сдвига в 4-угольном кольце промежуточного состояния, в котором электронная плотность почти выровнена. При этом протонированная карбоксильная группа после перегруппировки имеет такое же строение, как исходная, с тем лишь исключением, что содержит 18О вместо 16О.
Наши эксперименты не позволяют установить, имеет ли здесь место череда быстрых перегруппировок или же сольватный комплекс с водой представляет собой чистое промежуточное состояние, в котором электронная плотность обычных и водородных связей полностью выровнена. Однако независимо от этого состояние карбоксильной группы будет зафиксировано только тогда, когда лишний протон будет ею потерян (его унесет другая молекула вода в виде гидроксоний-катиона).
При этом лишь случайность определяет, с каким изотопом кислорода (16О или 18О) останется карбоксигруппа.
В реальных случаях на вероятность исхода оказывают влияние изотопная чистота применяемой воды H 2 18О (обычно всегда содержащей в себе остаточные примеси воды H216О) и соотношение молярных концентраций карбоксильного соединения и воды (поскольку в процессе обмена изотопная чистота воды H 2 18О падает, разбавляясь образующейся водой H 2 16О).
ВЫВОДЫ
Таким образом, на примере ряда пептидов продемонстрирована возможность в лабораторных условиях вводить в карбоксильные группы стабильный изотоп 18О и получать стандарты, пригодные для количественного анализа с использованием масс-спектрометрии.
Введенный в карбоксильные группы пептидов изотоп 18О сохраняется в водных растворах в широком диапазоне pH.
За исключением молекулярной массы, полученные изотопсодержащие соединения полностью сохраняют свои физико-химические характеристики, такие как хроматографическая и электрофоретическая подвижности. Это делает их идеальными стандартами не только при добавлении в пробы перед масс-спектрометрическим анализом, но и в процессе подготовки к анализу, поскольку позволяет исключить ошибки из-за возможной потери образца.
МАТЕРИАЛЫ И МЕТОДЫ
В работе использовали n-(тозил)аргинин (TA) фирмы Sigma; ангиотензин I, брадикинин, даларгин производства фирмы Sigma (Германия). Использовались данзил-глутамин и данзильное производное глутаминовой кислоты фирмы Reannal (Венгрия). Изотопный обмен проводили в H 2 18O производства B.O.C. Limited (Великобритания). Кроме того, использовались трифторуксусная кислота фирмы Sigma (Германия) и муравьиная кислота (Merck, Германия).
Хроматографирование проводили на микроко-лоночном жидкостном хроматографе Милихром А-02 (Эконова, Новосибирск). Использовалась хроматографическая колонка размером 2×75 мм, заполненная сорбентом Prontosil AQC18 (Bishof, Германия).
Масс-спектрометрические измерения проводили на времяпролетном масс-спектрометре МХ-5310, с ортогональным вводом ионов и оборудованном электрораспылительным источником ионов (ESI-o-TOF) (Институт аналитического при- боростроения РАН, г. Санкт-Петербург). Все спектры получены в режиме съемки положительных ионов. Объем пробы — 10–50 мкл, скорость подачи пробы — 1–5 мкл/мин.
МALDI масс-спектры были получены на приборе Ultraflex (Bruker Daltonics, Bremen, Германия). Все спектры получены в режиме съемки положительных ионов. В качестве матрицы использовалась α-циано-4-гидроксицинаминовая кислота.
Определение зависимости скорости обмена от температуры проводили в H 2 18O при температурах от 20 до 70 оС. К 50 мкл H 2 18O добавляли 3 мкл ТФУ. После чего полученную смесь делили на 5 порций по 10 мкл. К каждой пробе добавляли по 1 мкл раствора даларгина 20 мг/мл. Обмен проводили при температурах 20, 37, 50 и 70 оС в течение 30 мин.
После протекания реакции обмена пробу разбавляли в 10 раз. Очистку проводили хроматографически. Отобранную фракцию исследовали масс-спектрометрически.
Определение зависимости скорости обмена от концентрации ТФУ проводили в смеси трифторуксусной кислоты и H 2 18O. К 50 мкл H 2 18O добавляли 1.3, 1.5, 3, 6 и 12 мкл 100 % -й ТФУ соответственно. Для введения изотопной метки использовали даларгин, взятый в концентрации 2 мг/мл. Изотопный обмен проводили при t = 50 ºC в течение 30 мин. После протекания реакции обмена, пробу разбавляли в 10 раз в воде. Выделение даларгина проводили хроматографически. Отобранную фракцию анализировали на масс-спектрометре.
Для изучения стабильности метки в качестве субстрата использовали меченый ангиотензин I (Ang I). Реакции проводили в 12.5, 1.25 и 0.125 % растворах трифторуксусной и муравьиной кислот. Также использовались 12.5 и 1.25 % растворы уксусной кислоты. Смешивали 40 мкл раствора кислоты и 10 мкл смеси меченых стандартов. Обмен проводили при комнатной температуре (20 ºС) в течение 70 ч. Пробы отбирали через 23, 48 и 70 ч обмена. Реакцию останавливали путем разбавления реакционной смеси в 100 раз стандартным растворителем для масс-спектрометрии (50 % MeCN, 0.1 % AcOH). Соотношение включенных атомов кислорода определяли масс-спектрометрически на приборе МХ-5310 с ионизацией типа "электроспрей". В качестве меры изотопного соотношения меченых стандартов использовали степень включения метки n 18О.
Дезамидирование проводили в сильнокислой среде. В качестве субстрата использовали дансильное производное глутамина (Dan-Gln). Для дезмидирования использовали раствор Dan-Gln с концентрацией 1 мг/мл (2.64∙10–3 М). К 50 мкл раствора Dan-Gln добавляли 3 мкл ТФУ (конц., С(ТФУ) = 0.72 М) или 4.5 мкл муравьиной кисло- ты (конц., С0(МК) = 2.15 M). Реакцию проводили в термостате при 50 ºС в течение 6 ч. Соотношение исходного вещества (S) и продукта (Dan-Glu, P) контролировали с помощью ВЭЖХ и масс-спектрометрии (МС).
ВЭЖХ проводили на приборе Милихром А-02 при следующих условиях.
T = 35 ºC, V = 200 мкл/мин, подвижная фаза: A — 0.25 % ТФУ, H 2 O; Б — 0.25 % ТФУ, CH 3 СN; градиент А-Б 0–100 %, 10 мин.
Расчет степени включения изотопа 18О в пептиды (Σ18О) был произведен с помощью специального алгоритма (см. Приложение).
ПРИЛОЖЕНИЕ
-
I . Численные расчеты выполнены с помощью специальной программы, написанной нами для табличного редактора Microsoft Excel™ на встроенном языке VBA (Visual Basic Application), позволяющей обрабатывать табличные данные, продуцируемые программным обеспечением масс-спектрометра (Bruker Daltonics FlexAnalisys™). Расчетная часть состоит из двух последовательных этапов.
На первом этапе на основании интенсивностей линий изотопного мультиплета вычисляется гистограмма процентного содержания изотопных форм, отличающихся числом изотопов 18O в молекуле. Необходимое для расчета изотопное распределение для незамещенной формы может быть получено чисто вычислительным путем на основании известного содержания природных изотопов в земной коре или измерено экспериментально. Изотопные распределения меченых форм получаются из последнего посредством сдвига всего мультиплета в сторону высоких масс с шагом в две массовые единицы на каждый включенный изотоп 18O. Программа обеспечивает вычисление процентных вкладов (весов) каждой из этих форм, аппроксимируя экспериментальный спектр их суммой. Для дальнейших вычислений необходимыми являются только процентные содержания незамещенной формы как в меченом стандарте ( P s ), так и в смеси его с образцом ( P m ).
На втором этапе, вычисляется массовое отношение образца и стандарта в их смеси, исходя из следующих соображений. Поскольку образец в чистом виде не содержит изотопных форм, то их присутствие в смеси (100 – P m ) может быть обусловлено исключительно вкладом стандарта. Следовательно, вклад стандарта в незамещенную форму Pm может быть вычислен по формуле
P x = P s (100 – P m )/(100 – P s ), и тогда искомое отношение приобретет вид:
( P m – P x )/(100 – P m + P x ).
Полученное таким образом отношение массы образца к массе стандарта является достаточным для вычисления концентрации образца в анализируемой пробе, учитывая то, что концентрация стандарта является известной величиной.
Во многих случаях удовлетворительная точность может быть достигнута даже без измерения спектра меченого стандарта. В этом случае программа предсказывает величину вклада P s на основании параметров распределения вкладов замещенных форм, которое по теоретическим соображениям должно отвечать биномиальному закону. Программа подбирает параметры теоретического биномиального распределения, обеспечивающие наименьшую квадратичную ошибку. При этом величина P s получается интерполяцией.
Последний способ расчета сильно облегчает анализ природных белковых смесей, когда стандартом служит аналогичная смесь, подвергнутая изотопному обмену, а смешивание со стандартом предшествует дальнейшим процедурам разделения и очистки. В этом случае получение данных для чистого стандарта может оказаться весьма проблематичным, поскольку для этого потребовалось бы проведение параллельной работы по разделению меченой смеси. Кроме того, даже такие работы обычно не могут обеспечить должной точности количественного анализа из-за невоспроизводимых потерь вещества в ходе разделения. В этом случае дополнительный метод расчета, основанный на оценке параметров гистограммы распределения меченых форм, может оказаться незаменимым.
-
II.
а
б
^ х104
ф 25'
g 2-0'
- 1.5■
-
1 .0 ■
0.5 ■
5 ×10P го 1 " 5'
V)
Ф 4'
-
- 3■
2 ■
-
1 •
-
^ ×10P
га „
(/)
-
ф
-
"2 -
- 1 -
- 0.0P
m / z
678 680 682 684 686 688 690
m / z
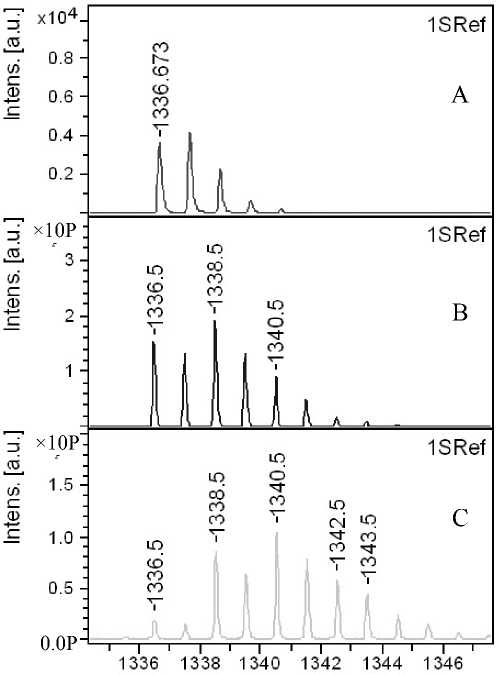
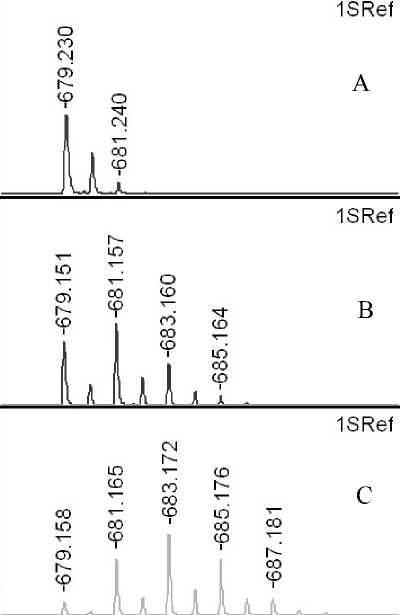
Рис. 1П. Фрагменты MALDI-MS спектров пептидов:
а — Р1 ( m / z 679.3) и б — Р2 ( m / z 1336.5) до обмена (A), после обмена в течение 50 мин (B)
и 100 мин (C)
а
m / z
б
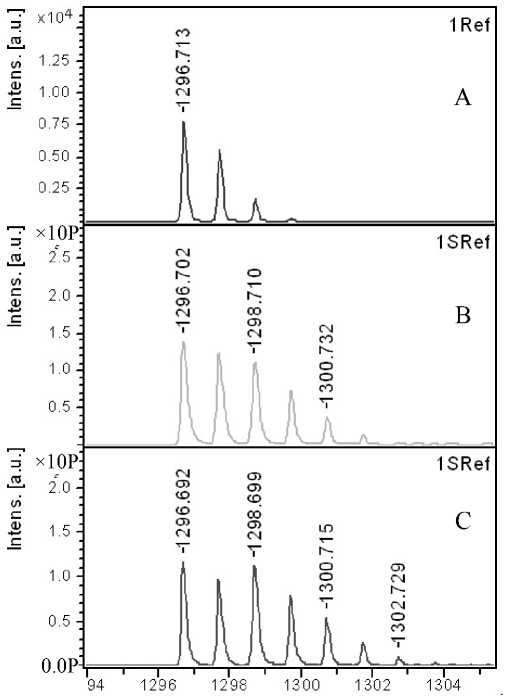
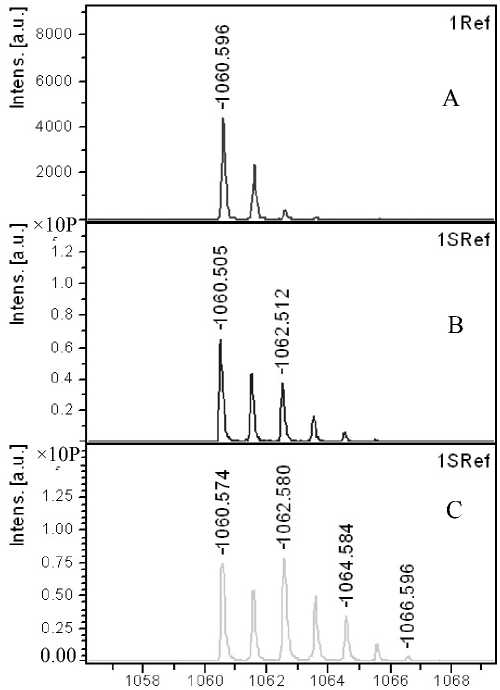
Рис. 2П. Фрагменты MALDI-MS спектров пептидов:
а — Br ( m / z 1060.6) и б — Ang I ( m / z 1296.7) до обмена (A), после обмена в течение 50 мин (B) и 100
мин (C)
m / z
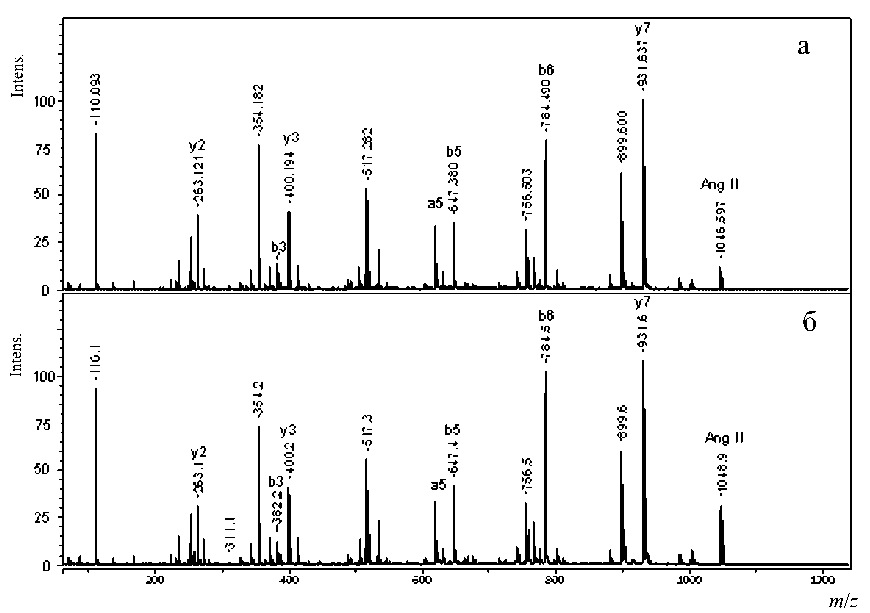
Рис. 3П. MALDI-MS/MS спектр Ang II после обмена в течение 50 мин (а) и 100 мин (б)
Intens. I n t ens .
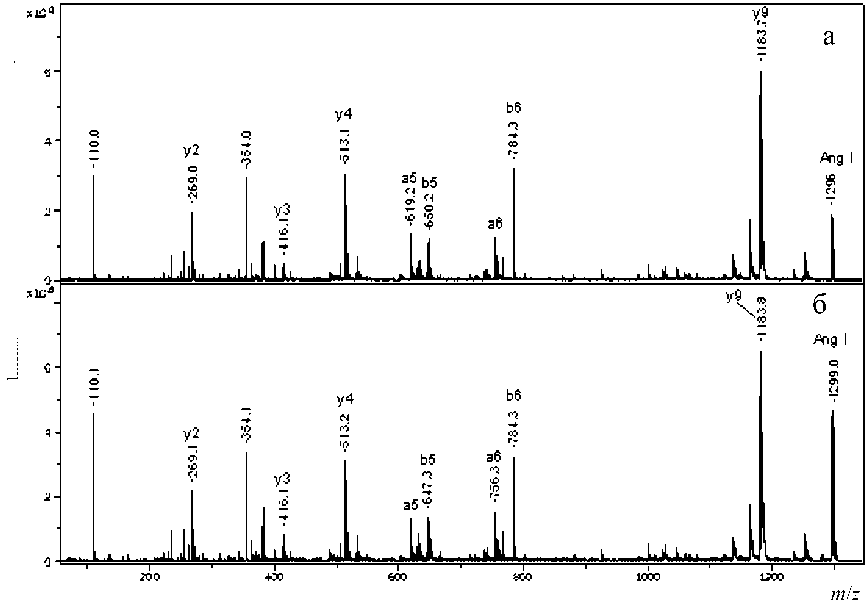
Рис. 4П. MALDI-MS/MS спектр Ang I после обмена в течение 50 мин (а) и 100 мин (б)
Intens. Intens. Intens.
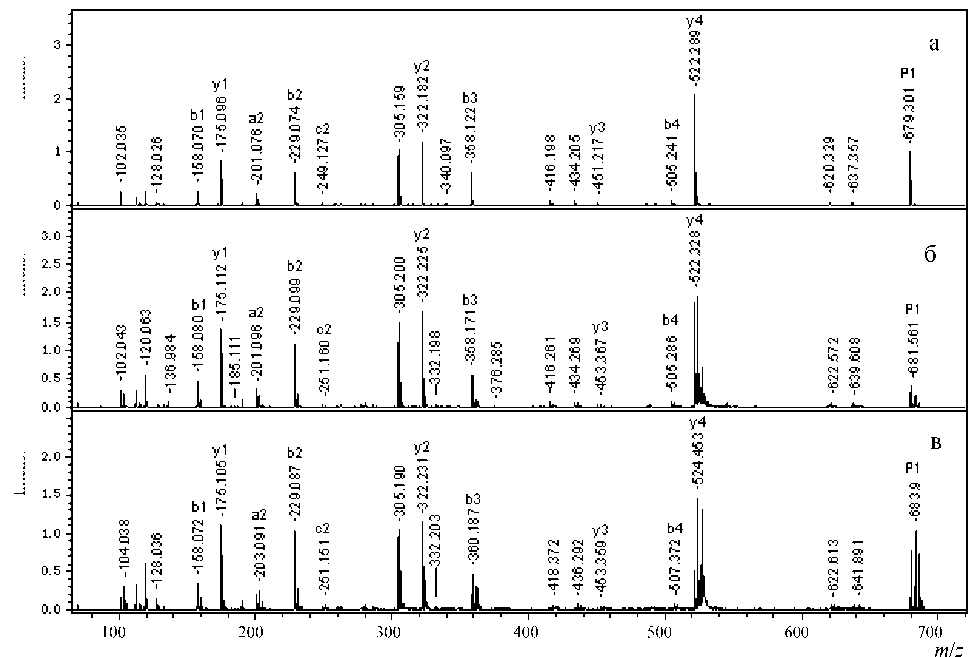
Рис. 5П. MALDI-MS/MS спектр Р1до (а), после обмена в течение 50 мин (б) и 100 мин (в)
а
б
в
m / z
Рис. 6П. Фрагменты MALDI-MS/MS спектров для отдельных ионов, содержащих остатки Glu (а), AcAsp (б) и Arg (в) до (А) и после обмена в течение 50(В) и 100 (С) мин
174 176 178 180 182
m / z
