Перманентный неонатальный сахарный диабет: клиническое наблюдение

Автор: Утц И.А., Шабаров В.К., Кравченя А.Р., Иванова С.Б., Новикова А.Н.
Журнал: Саратовский научно-медицинский журнал @ssmj
Рубрика: Педиатрия
Статья в выпуске: 2 т.11, 2015 года.
Бесплатный доступ
Цель: изучить особенности клинико-лабораторной картины неонатального сахарного диабета у недоношенного ребенка; провести анализ течения заболевания на протяжении 18 месяцев, определить механизмы развития неонатального сахарного диабета, разработать дифференцированный подход в терапии неонатального сахарного диабета.
Гликемический профиль, мутация хромосом, неонатальный сахарный диабет
Короткий адрес: https://sciup.org/14918108
IDR: 14918108
Permanent neonatal diabetes mellitus: clinical observation
The aim is to study the characteristics of clinical and laboratory picture of neonatal diabetes mellitus in a premature baby; to analyze the course of the disease for 18 months, to determine the mechanisms of development of neonatal diabetes mellitus, to develop a differentiated approach in the treatment of neonatal diabetes mellitus.
Текст научной статьи Перманентный неонатальный сахарный диабет: клиническое наблюдение
ных и перманентный СД. Частота выявления тран-зиторного НСД в популяции не превышает одного случая на 100–200 тыс. новорожденных. Перманентный НСД диагностируется менее чем в 5% случаев. Дети с транзиторной формой не требуют какого-либо лечения, так как уже к возрасту двенадцати недель симптомы диабета самопроизвольно исчезают. У них сохраняется высокая предрасположенность к развитию СД первого типа в более старшем возрасте, хотя клинических доказательств подобной закономерности не представлено. Перманентный НСД менее распространен, чем транзиторный НСД. Заболеваемость в среднем составляет 2,0 на 100 тыс. новорожденных. В 50% случаев ПНСД связан с мутациями SUR1 и Kir6.2 субъединиц КАТФ-канала. У всех пациентов отмечается внутриутробная задержка роста, в 80% случаев — диабетический кетоацитоз. Для них характерна гипертриглицеридемия, отсутствие секреции С-пептида и циркулирующих антигенов HLA DR3 и DR4. У всех пациентов в родословной отмечались родственные браки, а в семьях родственников аутоиммунные заболевания. Врожденное отсутствие островков с нормальной экзокринной тканью поджелудочной железы описано Dodge и Laurence. Темпы роста ускоряются после начала инсулиноте-рапии, что подтверждает роль инсулина в регуляции фетального роста. Перманентная форма СД требует назначения препаратов инсулина, так как самопроизвольно излечиться не может.
Многие научные исследования этой патологии основаны на единичных наблюдениях. Выявлен IPEX-синдром — крайне редкое заболевание, которое характеризуется сочетанием СД с хронической диареей, атрофией реснитчатого эпителия, клинически: экземой, анемией, тиреоидитом и рецидивирующей инфекцией. Данное заболевание обусловлено мутацией в гене FOXP3 (FOXP3-ген — Forkheadbox P3 scurfin, локализация — Xp11.23-q13.3).
В медицинском университете Сarol Davila (Romania) диагностирован синдромом Кернс–Сейра, KSS. KSS — это патология мышечной системы (митохондриальная миопатия) с офтальмоплегией, пигментной дегенерацией сетчатки, кардиопатией, СД и другими гормональными нарушениями.
В университете Josa Andras Hospital (Hungary) доктор I. Kantor описал пациента с мутацией гена BETA2/NeuroD, ответствененного за развитие мозговой ткани и поджелудочной железы. В результате его мутаций СД у детей развивается сахарный диабет, глухота, ретинопатия и нарушения координации.
F. Lombardo из Университета г. Мессина (Holy) описал семейный синдром Вольфрама с поражением сердечно-сосудистой системы. Полная форма DIDMOAD-синдрома обнаружена у нескольких детей (3 девочки и 4 мальчика). Все дети были рождены от близкородственных браков. При изучении гена WFS1 у всех пациентов обнаружена гомозиготная мутация в 8 экзоне.
P. Nakavachara (Университет г. Бангкока, Tail) описал сочетание DIDMOAD-синдрома с двусторонним гидронефрозом, гидроуретером и полиурией.
В Саратовской области под наблюдением детских эндокринологов находятся двое детей c неонатальным Сд при общей численности населения 2500000 человек.
Этиология. Причиной развития транзиторной формы СД новорожденных является импринтинг генов. Перманентная форма данной патологии развивается в ситуациях, когда имеет место наследственный дефект в гене, кодирующем АТФ-калиевый канал клеток поджелудочной железы, отвечающих за синтез инсулина. Неонатальный сахарный диабет вызван мутацией гена KCNJ11, который кодирует Kir6.2 субъединицы каналов KATФ бета-клеток поджелудочной железы. Мутации также могут происходить и в таких генах, как GCK, KCNJ11, INS и ABCC8. Вследствие нарушенного синтеза секреции инсулина во время внутриутробного периода происходит задержка развития плода, что при рождении проявляется недостаточным весом.
Именно малый вес при рождении и плохая прибавка веса заставляют родителей обратиться за помощью к специалистам, которые в ходе клинических и лабораторных исследований часто выявляют гипергликемию, метаболический кетоацидоз, глюкозурию и полиурию. При транзиторной форме СД обращают на себя внимание низкая масса тела, выраженная дегидратация ребёнка за счёт полиурии, нарушение кислотно-основного равновесия и несоответствующая возрасту масса тела.
Клиническое наблюдение. В марте 2012 г. в ГУЗ «Перинатальный центр» г. Саратова от 4-й беременности, 2-х срочных родов путем операции кесарева сечения по поводу угрожаемой асфиксии плода родилась недоношенная девочка. Беременность про-
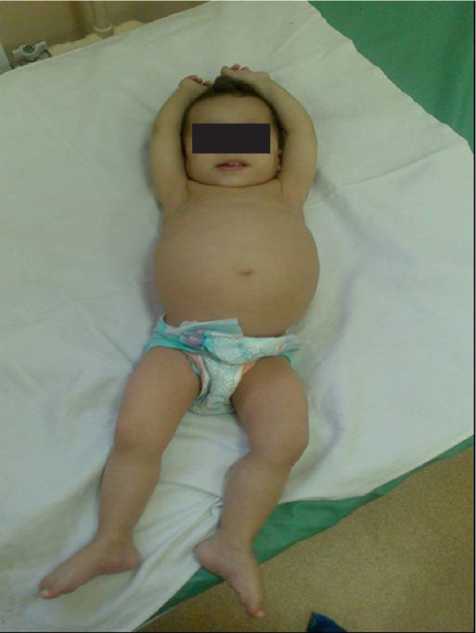
Рис.1. Ребенок с неонатальным сахарным диабетом (возраст 18 месяцев)
текала на фоне отягощенного акушерского анамнеза (анемия, ХВУГП, гестоз) и сопровождалась обострением атопического дерматита. Родословная ребенка: не исключен близкородственный брак. Срок гестации: 36 недель. Масса при рождении 1520 г, рост 38 см, окружность головы 29 см, окружность грудной клетки 26 см. Состояние после рождения тяжелое за счет дыхательной недостаточности и морфофункциональной незрелости. В ОРИТН находилась до 9-х суток жизни, на СРАР в течение двух суток, затем была переведена в отделение патологии новорожденных ГУЗ «Перинатальный центр», позже в ОРИТ ГУЗ «СОДКБ». С июня 2013 г. по настоящее время находится на лечении в эндокринологическом отделении ГУЗ «СОДКБ» (рис. 1).
При проведении неонатального скрининга пять врожденных заболеваний: врожденный гипотиреоз, муковисцидоз, адреногенитальный синдром, галактоземия, фенилкетонурия — были исключены. У девочки с первых часов рождения колебания гликемии составили от 2,5 до 40 ммоль/л. В ГУЗ «Перинатальный центр» после введения 1 ед инсулина (гликемия 40,0 ммоль/л) ее уровень снизился до 0,8 ммоль/л дальнейшая динамика (через 6-9-12 часов) уменьшения гликемии до 0,5–1,8 ммоль/л. При поступлении гипергликемия 30,9–35,4,41,6 ммоль/л, глюкозурия 0,5-1,6%, кетоацидоз отсутствовал. Старт лечения с инсулинов короткого и ультракороткого действия, но при минимальных дозах 0,01–0,02 ед. у пациентки отмечались частые тяжелые гипогликемические состояния. Препаратом выбора определен левемир — инсулин пролонгированного действия с плоским профилем активности. В ОРИТ «СОДКБ» обращали на себя внимание: нестабильные цифры гликемии, задержка физического и психомоторного развития, синдром мальабсорбции, крайне лабильное течение заболевания и очень высокая чувствительность к минимальным дозам инсулина.
Проведено генетическое исследование в ФГУЗ «ЭНЦ» (г. Москва): известные генетические нарушения по данным прямого секвенирования и анализа кодирующей последовательности гена КСNJ11 (Kirb.2) не выявлены. Попытка использования препаратов сульфанилмочевины не увенчалась успехом. В настоящее время ферментативная недостаточность и синдром мальабсорбции ухудшают течение диабета. Колебания гликемии за сутки могут составлять 1,8–35,5–42,3 ммоль/л, чаще в пределах 8,2–22,4. Дважды отмечены эпизоды гипогликемической комы (в мае 2013 г. и 31 марта 2014 г.) с клиникой нарушения мозгового кровообращения, которое купировалось в течение пяти часов. Изложенное отражает увеличение потребности в инсулине, доза которого составляет от 0,5 до 2,5 ед/сут. Настоящая гликемия чаще на цифрах 5,29–11,8 ммоль/л.
Объективные данные. Состояние при поступлении: средней тяжести за счет метаболических нарушений, декомпенсации углеводного обмена и перинатального поражения ЦНС. Положение естественное. Тепло удерживает. Телосложение астеническое. Кожные покровы: чистые, смуглые. Гипертрихоз. Влажность кожи достаточная. Эластичность кожи снижена. Слизистые чистые, бледно-розовые, влажные. Подкожно-жировой слой развит слабо. Имеются участки постинъекционых гиперлиподи-строфий на бедрах, ягодицах. Тургор тканей несколько снижен. Мышечный тонус снижен. Видимых отеков нет. Лимфатические узлы не увеличены. Дыхание: спонтанное, адекватное, ЧДД 38 в минуту, одышки нет. Дыхание пуэрильное. Сердечно-сосудистая система: область сердца не изменена. Границы относительной сердечной тупости соответствуют возрасту: левая на 0,5 см кнаружи от левой среднеключичной линии, правая — правый край грудины, верхняя — второе ребро. Верхушечный толчок ограниченный, средней силы и интенсивности, локализация в 4-м межреберье на 0,5 см кнаружи от левой среднеключичной линии. Тоны сердца ясные, ритмичные. Шум систолический, низкой интенсивности, на верхушке. Пульс: ритмичный, удовлетворительного напряжения и наполнения, одинаков на обеих руках. АД 75/35 мм. рт. ст. Кисти, стопы теплые, симптом «белого пятна» 3 секунды, ЧСС 130–140 в минуту.
Аппетит сохранен. Кормление через рожок, усваивает. Срыгивания нет. Глотание свободное. Живот: увеличен в объеме. Пупочная грыжа. Перистальтика кишечника выслушивается во всех отделах. Печень выступает на 1,5–2 см из-под края реберной дуги. Селезенка не увеличена. Щитовидная железа: не пальпируется. Половое развитие: наружные половые органы диспластичны, большие половые губы не прикрывают малые. Развиты по женскому типу. Ma0, P0, Ax0, Me0. Стул до 5–9 раз в сутки, кашицей, с непереваренными комочками. Мочится свободно, диурез и жажда значительно увеличиваются при повышении гликемии (стойком) выше 25 ммоль/л. Неврологический статус: крик громкий, реакция на осмотр адекватная. Поза полуфлексии. Физиологические рефлексы вызываются. Глубокие рефлексы D=S понижены. Чувствительность не нарушена.
Динамика клинических данных . За время наблюдения в ГУЗ «СОДКБ» (временной промежуток с 06.2012 по 02.2014) отмечается положительная ди-
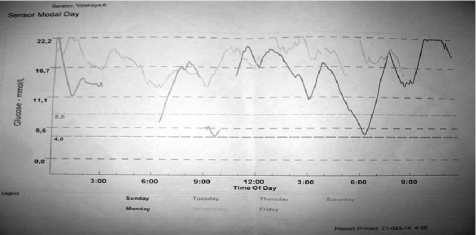
Рис. 2. Суточное мониторирование глюкозы СGMS: 1-е сутки наблюдения
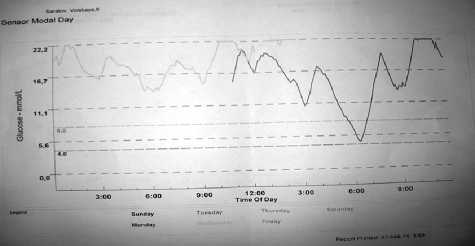
Рис. 3. Суточное мониторирование глюкозы СGMS: 3-и сутки наблюдения намика в общем состоянии ребенка. Телосложение астеническое. Липодистрофии: гипертрофия на плечах, бедрах (осложнения инсулинотерапии). Суставы: безболезнены, не изменены. Мышечная система развита слабо. Тонус мыщц снижен. Зубы 4/2. Живот: вздут, увеличен в объеме. Печень: на 1,5 см из-под края реберной дуги. Аппетит повышен. Диурез повышен, адекватен вводимой жидкости. Психомоторное развитие с задержкой. Стоит в кровати, ходит неуверенно с поддержкой за обе руки несколько шагов. Речь не развита, повторяет слоги. Вес 6,65 кг (плюс 6 кг за 24 месяца). Рост 67,0 см, ИМТ 14,81 кг/м 2, SDS –4,6.
Физическое развитие: соответствует возрасту 6 месяцев (п/ц). Половое развитие: Ma0, P0, Ax0, Me0, гипертрофия клитора.
Лабораторные исследования: ОАК — анемия HB — 93 г/л, анизоцитоз и пойкилоцитоз 1-й степени. ОАМ — глюкозурия (0,3-2,6%), микропротенурия 0,02–0,08 г/л.
Гликемический профиль: 15,0–32,8–39,6–6,2–9,3 ммоль/л (рис. 2, 3).
С-пептид: 1522 н/ммоль (норма 298–1324).
С-пептид через 2 часа 1622 нмоль/л. Иммунореактивный инсулин 0,0–0,6 мкмоль/мл.
Антитела к инсулину 2,5 ед/мл (норма до 5), в динамике 6,4 ед/мл.
Проинсулин 2,6 пмоль/л (норма 3,3-28).
Гликированный гемоглобин НЬА114,1-16,5-9,3%. АКТГ 15,4 пг/мл (норма 8,3–57,8).
Кортизол 7:00505 нмоль/л (норма до 660).
ТТГ 3,9 Мкед/мл (норма 0,5–4,0).
Св.Т415,9 пмоль/л (норма 10-23,2).
АТ ТПО — отрицательный результат. Ренин 2,6 мкМЕ/мл (4,4–46,1).
Белковые фракции: альбумины: 64%, глобулины общие: 36%, альфа: 10%, бетта: 10%, гамма: 16%.
Электролиты: натрий ионизированный: 131,0 ммоль/л. Калий ионизированный: 5,24 ммоль/л. Кальций ионизированный: 1,18 ммоль/л.
КОС — рН: 7,360; PCO2; 46 mmHg; PO2 66 mmHg, HCO3 act: 27,4 ммоль/л; HCO3 Std: 26 ммоль/л, BE (ect) 0,0 ммоль/л BE, (B): 0,4 ммоль/л, tCO2: 24.8 ммоль/л, O2 SAT: 91,7, O2 (CT): 14,6 mL/dL, tHb (расч): 11,3 г/дл, Na+: 132.4 ммоль/л, Ka+: 6,12 ммоль/л, Hct: 34%, Cl-: 93,8 ммоль/л.
АТ к ВИЧ, HBs-Ag, антитела к HCV не обнаружены.
ЭКГ — лёгкая, синусовая, аритмия ЧСС 120–136 уд. в минуту. Срединное положение ЭОС.
УЗИ внутренних органов: размеры печени увеличены (среднезадний размер правой доли 81 мм, левой доли 57 мм). Реактивные изменения поджелудочной железы. Эхо-признаки обменных изменений паренхимы почек. Определяются единичные однородные мезентериальные лимфатические узлы размером до 5–10 мм.
УЗИ вилочковой железы: признаки тимомегалии: правая доля 33*18*30мм, левая доля 35*16*30мм.
Консультация невролога: энцефалопатия, синдром двигательных нарушений, острое нарушение мозгового кровообращения в анамнезе.
Консультация гастроэнтеролога: реактивный гепатит, синдром мальабсорбции.
Консультация ортопеда: дисплазия тазобедренных суставов.
Консультация офтальмолога: диагноз: фоновая ретинопатия.
Диагностика. Наша пациентка и все новорожденные, находящиеся в критическом состоянии, должны быть в обязательном порядке обследованы для исключения НСД. Так как у нее в течение первых шести месяцев жизни СД, в обязательном порядке проведено генетическое тестирование на мутации, связанные с АТФ-калиевым каналом, и сделан анализ полиморфизмов в генах ADAMTS9, JAZF1, KCNJ11, KCNQ1, PPARG, TCF7L2.
Таким образом, у нашей пациентки выставлен основной клинический диагноз: неонатальный сахарный диабет наследственного генеза.
Осложнения: острое нарушение мозгового кровообращения в анамнезе, гипогликемические комы в анамнезе (05.05.13, 31.03.14).
Сопутствующие: дистрофия с дефицитом массы тела 1-й степени, микросомия, гипертензионно-гидроцефальный синдром, тонусные нарушения, анемия легкой степени тяжести, тимомегалия, реактивный гепатит, синдром мальабсорбции, пупочная грыжа, дисплазия тазобедренных суставов, фоновая ретинопатия.
Лечение. Известны случаи успешной коррекции состояния ребёнка при перманентной форме СД новорожденных без применения инсулина, благодаря назначению препаратов сульфанила мочевины. У пациентов с НСД, несмотря на применение инсулина, трудно достичь эугликемии. Однако инсулин является единственным препаратом, позволяющим добиться коррекции состояния. При транзиторной форме НСД необходимо восстановить кислотно-основной и водно-электролитный баланс. В настоящее время наша пациентка получает инсулиноте-рапию: актрапидп/к: 6.000,05-0,1 ед., 9.00-0,15 ед., 13.300,15 ед., 18.00-0,15 ед., 21.300,1 ед. по гликемии и в зависимости от углеводного коэффициента пищи. Левемир п/к: 9.000,6-2 ед., 21.000,05 ед. Также проведены: гепатопротекция (гептрал, хофитол); ферментотерапия (креон); профилактическая проти- вовирусная терапия (виферон, гриппферон); 2 курса ноотропной терпии.
Заключение. Прогноз зависит от формы данной патологии и сроков её выявления. При условии своевременной коррекции нарушений гомеостаза транзиторная форма СД новорожденных обещает благоприятный прогноз, а её перманентный вариант предполагает пожизненное применение инсулина с ранним развитием сосудистых осложнений.
Список литературы Перманентный неонатальный сахарный диабет: клиническое наблюдение
- Баррет Т.Г. Генетические синдромы и сахарный диабет. Детский диабет 2006; (7): 3-10
- Карлсон А. Лучшая диагностика диабета: Регистрация случайных пациентов по всей стране предполагает изменение распределения генотипа HLA (антигены лейкоцитов человека) с 1986-1987 г. Детский диабет 2006; (7): 27-30
- Галли-Цинополу. Аллельный ген HLA у детей и подростков с сахарным диабетом 1 типа в Северной Греции. Детский диабет 2006; (7): 17-20
- Кантор И. Клинические характеристики пациента, страдающего от бета-2-генетической патологии. Детский диабет 2006; (10): 19-25
- Миху M. Синдром Керне -Сейра: Задержка диагностики в одном из случаев. Детский диабет 2006; (8): 15-25
- Паскова M. Сульфонилмочевина вместо инсулина у PNDM пациентов с активацией мутации в гене KCNЛ1 кодирования Kirb 2 субъединицы привело к значительному улучшению компенсации диабета. Детский диабет 2006; (8): 34-40
- Перец А. Генетические и иммунологические основы диабета в многочисленной единокровной семье бедуинов. Детский диабет 2006; (9): 34-39
- Рами Б. Клинические и молекулярные находки в нетипичном случае IPEX-синдрома (Синдром иммунной диерегуляции, полиэндокринопатии, энтеропатии. Детский диабет 2007; (7): 28-32
- Скорка А. Анализ генетических факторов, предрасполагающих к возникновению диабета 1 типа у детей в возрасте до 5 лет. Детский диабет 2007; (7): 25-30
- Слингерланд А. Влияние мутаций и лечения на диабет и неврологию. Детский диабет 2007; (7): 30-40
- Датц H. Клинические параметры молекулярного тестирования зрелости диабета у молодых. Детский диабет 2007; (8): 52-56
- Джессик M. Зрелость-развитие диабета молодых, или MODY 3 типа: описание случая ребенка женского пола, восприимчивого к низкой дозе сульфонилмочевины. Детский диабет 2007; (8): 53-60
- Хофер С. Семейный отчет брата и сестры, страдающих переходным диабетом новорожденных с наследуемой от отца новой мутацией рецептора 1 сульфонилмочевины. Детский диабет 2007; (8): 58-65
- Ломбардо Ф. Участие сердечно-сосудистой системы в синдроме Вольфрама. Детский диабет 2007; (8): 50-57
- Нокавачара П. Мальчик с синдромом Вольфрама, с сахарным диабетом и признаками полиурии, двустороннего гидронефроза и гидроуретера (аномального растяжения мочеточника с мочой), имитирующего обструктивную уропатию. Детский диабет 2007; (8): 51-56
- Микле И. Постоянный сахарный диабет новорожденных -терапия сульфонилмочевиной. Детский диабет 2007; (8): 55-60.
