Первый случай пренатальной диагностики мукополисахаридоза IV типа (синдром Моркио) в Азербайджанской Республике
 в Азербайджанской Республике")
Автор: Ализаде Севда Айдын, Алиева Камиля А., Мамедбейли Айтен Камал, Мусаев Ширхан Тариел, Расулов Эльхан М.
Журнал: Бюллетень науки и практики @bulletennauki
Рубрика: Медицинские науки
Статья в выпуске: 11 т.8, 2022 года.
Бесплатный доступ
Во время экспедиционных работ по выявлению больных с мукополисахаридозом в одном из регионов Азербайджанской Республики при клиническом обследовании врача-педиатра, врача-невролога и врача-генетика выявлена девочка пяти лет с подозрением на мукополисахаридоз. Для установления типа мукополисахаридоза использовали следующие лизосомальные ферменты: альфа-L-идуронидаза (МПС I), идуронат-2-сульфатаза МПС II), гепарин N-сульфатаза (МПС IIIA), альфа-N-ацетилглюкозаминидаза (МПС IIIB), ацетил-КоА-глюкозамин-N-ацетилтрансфераза (МПС IIIC), N-ацетилглюкозамин-6-сульфатаза (МПС IIID), N-ацетил-галактозамин-6-сульфатаза (МПС IVA), бета-галактозидаза (МПС IVВ) и арилсульфатаза B (МПС VIВ). Анализ активности фермента N-ацетил-галактозамин-6-сульфатаза показал низкую активность 0,1 μmol/L при норме >0,2 μmol/L/h, что характерно для гомозиготного или двойного гетерозиготного состояния синдрома МПС IVA типа. Молекулярно-генетический анализ гена GALNS идентифицировал замену нуклеотида аденин на нуклеотид гуанин в позиции 1283 (с.1283 A>G). Известно, что в следствие мутации на уровне белка происходит замена аминокислоты глицин на аргинин в позиции 428, Gln428Arg (Morquio A.; OMIM®: 253000). Дополнительное исследование членов семьи (родителей и сестры) пробанда показали гетерозиготное состояние выявленной мутации. В момент обследования мать пробанда была беременной. На 16-17-недельном сроке беременности произвели трансабдоминальный амниосентез. На уровне ДНК, выделенной из фибробластов амниотической жидкости, установили гетерозиготное носительство мутации с.1283 A>G гена GALNS, что свидетельствует о здоровом плоде. Родители были ознакомлены с результатами анализа и по согласию обоих родителей плод был сохранен для дальнейшего развития. Планируем проведение пренатальной диагностики всем семьям репродуктивного возраста с наличием больных детей с диагнозом МПС.
Мукополисахаридоз iva типа, синдром моркио, n-ацетил-галактозамин-6-сульфатаза, фермент, ген galns, мутация, белок, пренатальная диагностика
Короткий адрес: https://sciup.org/14126203
IDR: 14126203 | УДК: 616-008.9-056.7:616.71 | DOI: 10.33619/2414-2948/84/41
First case of prenatal diagnostics of type IV mucopolysaccharidosis (Morquio syndrome) in Azerbaijan Republic
In one of the regions of Azerbaijan Republic while clinic examination with doctor-pediatrician, doctor-neurologist and doctor-geneticist a girl of 5 years of age was identified as suspicious of mucopolysaccharidosis. N-acetyl-galactosamin-6-sulfatase enzyme analysis has shown low activity of 0,1 μmol/L at norm of >0,2 μmol/L/h. Molecular-genetic analysis of GALNS gene identified substitution of Adenine nucleotide with Guanine nucleotide in 1283 (с.1283 A>G) position accompanied with amino acid change Gln428Arg (Morquio A; OMIM®: 253000). These results correspond to the presence of MPS IVA type. With the consent of both parents and at the term of 16-17-week pregnancy transabdominal amniocentesis was carried out. At the level of DNA, isolated from fibroblasts from amniotic liquid, we have got heterozygous carriage of a с.1283 A>G mutation of GALNS gene, that says of a healthy fetus. We plan to carry out prenatal diagnostics to all families of reproductive age, who have got affected children diagnosed with MPS.
Текст научной статьи Первый случай пренатальной диагностики мукополисахаридоза IV типа (синдром Моркио) в Азербайджанской Республике
Бюллетень науки и практики / Bulletin of Science and Practice
УДК 616-008.9-056.7:616.71
Мукополисахаридозы (МПС) — группа наследственных болезней обмена веществ, связанных с нарушением метаболизма гликозаминогликанов (ГАГ), приводящим к поражению органов и тканей. Мукополисахаридоз IV типа (МПС IV) (синонимы: болезнь Моркио, спондило-эпифизарная дисплазия, хондроостеодистрофия, деформирующая остеохондродистрофия, Моркио-Брайлсфорда синдром, Моркио-Ульриха синдром, эксцентрохондроплазия, Дугве-Мелхиора Клаузена синдром). Заболевание описано в 1929 году уругвайским педиатром Luis Morquio (1867-1935) и Джеймсом Фредериком Брэйлсфордом (1888-1961), английским рентгенологом в Бирмингеме, Англия [1-5].
МПС IV типа обусловлен дефицитом лизосомных гидролаз: N-галактозамин-6-сульфатсульфатазы (МПС IVА) или β-галактозидазы (МПС IVВ), отложением в соединительной ткани кератансульфата и характеризуется значительной деформацией скелета и отставанием в росте. Все вышеперечисленные признаки приводят к инвалидизации, а при тяжелом течении болезни - к летальному исходу. В отличие от других типов МПС, МПС IV типа характеризуется отсутствием снижения интеллекта, помутнения роговицы, гепатоспленомегалии. Различают несколько форм мукополисахаридоза IV типа: тяжелую, классическую, промежуточную и легкую. Ген N-ацетил-галактозамин-6-сульфатазы (GALNS) картирован на 16 хромосоме в локусе 16q24.3. Тип наследования МПС IV типа-аутосомно-рецессивный [6, 7].
Синдром Моркио — один из самых редких видов нарушений мукополисахаридов. Точных данных нет, но данное заболевание встречается приблизительно у 1 на 200000300000 здоровых новорожденных. И хотя данное заболевание - очень редкое, каждый пациент нуждается в столь обширном и разноплановом медицинском обслуживании. В США частота МПС всех типов составила 0,98 на 100 000 новорожденных в Польше 1,8 на 100 000, в Японии и Швеции 1,53 на 100 000, в Швейцарии 1,56 на 100 000 живых детей [8-12].
Впервые нами проведено эпидемиологическое исследование болезни МПС в Азербайджанской Республике. Исследование проводили за период 2018-2022 гг. Проведенный скрининг ферментного анализа из 56 подозреваемых больных у 26 пациентов выявил дефицит фермента N-ацетил-галактозамин-6-сульфатаза характерный для MПС IVA типа, что составил 46,4% от всех обследованных пациентов [13-14].
Дальнейшая наша цель разработать пути профилактики данного заболевания в виде медико-генетической консультации семей репродуктивного возраста c генетическим риском для последующей пренатальной диагностики плода.
В данной статье приводим результаты диагностики МПС IVA у пятилетней девочки. В момент идентификации типа мутации мать больной была беременной, что позволило нам воспользоваться ситуацией и с согласия супружеской пары провести пренатальную диагностику.
Материалы и методы
Во время экспедиционных работ в одном из регионов Республики в Центральной районной больнице выявлена девочка 5 лет с явными клиническими проявлениями характерными для болезни МПС. Образец крови больной был помещен на DBS (Dry Blood Sample) карту.
Биохимический анализ для дифференциальной диагностики мукополисахаридоза использовал следующие лизосомальные ферменты для всех типов МПС: альфа-L-идуронидаза (MПС I), идуронат-2-сульфатаза (MПС II), гепарин-N-сульфатаза (MПС IIIA), альфа-N-ацетилглюкозаминидаза (MПС IIIB), ацетил-КоА-глюкозамин-N-ацетилтрансфераза (MПС IIIC), N-ацетилглюкозамин-6-сульфатаза (MПС IIID), N-ацетил-галактозамин-6-сульфатаза (MПС IVA), бета-галактозидаза (МПС IVВ) и арилсульфатаза B (MПС VIВ).
Для определения активности ферментов использовали метод флюориметрии, а тестирование мутации проводили методом NGS (Next Generation Sequencing -Секвенирование нового поколения). ДНК, полученную из образца периферической крови пациента, исследовали методом секвенирования нового поколения. «Более 99% кодирующих областей этих генов были изучены с глубиной чтения не менее 50X. Средняя глубина чтения составляет 1559 показаний. В анализ были включены соединения экзон-интрон (±10 п.н.).
Классификацию патогенности полученных данных проводили согласно «Руководству ACMG*».
Забор амниотической жидкости проводили трансабдоминальным способом при сроке беременности 16-17 недель.
Результаты собственных исследований
Больная В. Г. девочка 5 лет с подозрением на болезнь MПС выявлена во время экспедиционных исследований. Больная была выявлена при клиническом осмотре врача-педиатра, врача-невролога и врача-генетика. При генеалогическом изучении у пробанда была младшая сестра трех лет, у которой отсутствовали клинические проявления МПС. Во время медико-генетического консультирования больной В.Г. мать была беременна. Воспользовавшись благоприятным моментом и известив об этом супружескую пару, был получен положительный ответ на пренатальную диагностику.
Родители — близкие родственники: двоюродные брат и сестра. Бабушки больной являются родными сестрами. Для всех членов семьи проведен анализ на определение активности лизосомальных ферментов для диагностики типа МПС: МПС I, МПС II, МПС III, МПС IV и МПС VI. Результаты ферментного анализа представлены в Таблице 1.
Таблица 1
РЕЗУЛЬТАТЫ АКТИВНОСТИ ФЕРМЕНТОВ ПО ТИПАМ МПС ДЛЯ БОЛЬНОЙ В.Г.
|
Фермент |
Активность |
Единица |
Норма |
|
альфа-L-идуронидаза |
17,6 |
μmol/L/h |
>1,5 |
|
идуронат-2-сульфатаза |
8,8 |
μmol/L/h |
>2,5 |
|
N-ацетил-галактозамин-6-сульфатаза |
5,1 |
μmol/L/h |
> 0,5 |
|
N-ацетилглюкозамин-6-сульфатаза |
0.1 |
μmol/L/h |
> 0,2 |
|
бета-галактозидаза |
1,2 |
μmol/L/h |
> 1,0, |
|
арилсульфатаза B |
10,9 |
μmol/L/h |
> 5,0, |
Анализ активности фермента N-ацетил-галактозамин-6-сульфатаза из высушенных пятен крови больной В.Г. показал низкую активность 0,1 μmol/L при норме >0,2 μmol/L/h, что характерно для синдрома МПС IVA типа. Уровень фермента был сильно снижен, что соответствует гомозиготному или двойному гетерозиготному состоянию заболевания. У родителей и у сестры пробанда уровень активности фермента N-ацетил-галактозамин-6-сульфатаза был снижен почти на половину от нормальной активности фермента (<0,7μmol/L/h-<0,17μmol/L/h), что характерно для гетерозиготного состояния дефицита фермента. Результаты ферментного анализа представлены в Таблице 2.
Tаблица 2
РЕЗУЛЬТАТЫ АНАЛИЗА ФЕРМЕНТА N-АЦЕТИЛ-ГАЛАКТОЗАМИН-6-СУЛЬФАТАЗА
У ЧЛЕНОВ СЕМЬИ ПРОБАНДА В.Г.
|
Пациенты |
Результаты |
Норма |
Зиготность |
Интерпретация |
Метод |
|
В.Г. |
<0,1(LOD) μmol/L/h |
≥2,0 mol/L/h |
Гомозигота |
Патогенный класс 1 |
Жидкостная хроматография |
|
Мать |
(<0,7 (LOD) μmol/L/h |
≥2,0 mol/L/h |
Гетерозигота |
Патогенный класс 1 |
Жидкостная хроматография |
|
Отец |
<1,0 (LOD) μmol/L/h |
≥2,0 mol/L/h |
Гетерозигота |
Патогенный класс 1 |
Жидкостная хроматография |
|
Сестра |
<0,9 (LOD) μmol/L/h |
≥2,0 mol/L/h |
Гетерозигота |
Патогенный класс 1 |
Жидкостная хроматография |
Для уточнения диагноза всем членам семьи проведена генетическая диагностика на уровне гена GALNS. Идентифицирована мутация - замена нуклеотида аденин на нуклеотид гуанин в позиции 1283 (с.1283 A>G). Следствием мутации на уровне белка происходит следующая аминокислотная замена: замена глицина на аргинин в позиции 428, Gln428Arg (Morquio A.; OMIM®: 253000).
Выявлялись случаи МПС IVА типа у населения Азербайджанской Республики и ранее. Больные отбирались при клиническом осмотре врача-педиатра, врача-невролога и врача-генетика. Проведенный скрининг и ферментный анализ для 26 пациентов из 56 помог выявить дефицит фермента N-ацетил-галактозамин-6-сульфатаза, характерного для MПС IVA типа, частота которого составила 46,4% от всех обследованных пациентов с диагнозом МПС, что говорит о преобладании над остальными типами заболевания. Ранее в наших исследованиях удалось идентифицировать мутацию 1283A>G и дополнительно шесть типов мутаций, которые имели только нуклеотидные замены: 553C>T, 439T>A, 157G>A, 463G-T, 1018G-T и 443A>G. Эти мутации были в гомозиготном, двойном гетерозиготном (компаунд) и в гетерозиготном состояниях [13-14].
Выявленные мутации гена GALNS у пациентов нашей Республики ранее обнаруженны и описаны в литературе. В нашей Республике МПС IVA типа занимает первое место среди всех типов МПС. Анализ литературы показывает иную картину: МПС IVA в популяциях Европейских и Азиатских стран занимает 3-4 места среди больных с диагнозом МПС [13-14].
Wang с соавт., (2010) при обследовании 24 больных китайcкой национальности выявили 27 различных типов мутации гена GALNS, 15 из которых были новыми мутациями [15].
Khan с соавт. (2017) при обследовании жителей Японии и Швеции за период 1982-2009 гг) выявили 469 больных с диагнозом МПС. Частота МПС IVА составил 10% от всех больных с диагнозом МПС. Эпидемиологические исследования больных МПС из Швейцарии за 34 года (1975-2008) выявлен 41 больных, частота которых составила 1,56:100000 живых новорожденных. Доля МПС IVА составил 24% от всех больных с диагнозом МПС [18].
В Британской Колумбии за период с 1952 по1986 гг выявлен один больной с диагнозом MПС IVA из 216 412 практически здоровых новорожденных [17].
Nelson с соавт., (2003) в Западной Австралии при обследовании 640 000 здоровых новорожденных за период 1969-1996 гг. выявлен только один случай рождения больного ребенка с диагнозом MПС IVA [16].
Caciotti с соавт. (2015) изучили 37 итальянских пациентов с MПС IVA и обнаружили, что стандартные процедуры секвенирования не смогли охарактеризовать вторую мутацию, вызывающую заболевание, у 16% пациентов. Поиск больших перестроек и дефектов мРНК в этих 16% выявил дефекты сплайсинга или большие делеции на другом аллеле в 67% из них [19]. Авторы сообщили о 14 новых мутациях в GALNS среди 37 пациентов. Анализируя литературные данные и сравнивая их с результатами по Республике, очевидно видим высокую встречаемость синдрома МПС, в целом, и MПС IVA, в частности. За период с 20182022 гг. по результатам наших исследований выявлено 52 пациента с гомозиготной, двойной гетерозиготной и гетерозиготной формой МПС. 46,4% из этих больных пришлись на долю МПС. Одним и основным объяснением высокой частоты встречаемости синдрома МПС среди населения Азербайджанской Республики является наличие высокой частоты кровнородственных браков, в частности: кровнородственных браков двоюродного типа.
Учитывая высокую встречаемость синдрома МПС и наличие ферментного и молекулярно-генетических анализов, единственным и правильным путем борьбы с генетической болезнью является профилактика болезни, включающая медико-генетическое консультирование с последующей пренатальной диагностикой плода в семьях с генетическим риском рождения больного ребенка.
У пробанда В.Г. с установленным диагнозом МПС IVA типа в момент обследования мать была на 16-й неделе беременности. Проведя просветительскую работу с родителями пробанда В.Г., получили согласие на проведение внутриутробной диагностики плода. Учитывая срок беременности, мы выбрали амниоцентез путем трансабдоминальной аспирации амниотической жидкости. На уровне ДНК, выделенных из фибробластов амниотической жидкости, провели генетическую диагностику плода. Параллельно для точности получаемых результатов провели генетический анализ на уровне ДНК, выделенных из лимфоцитов периферической крови родителей. Результаты представлены на Рисунках 1-3.
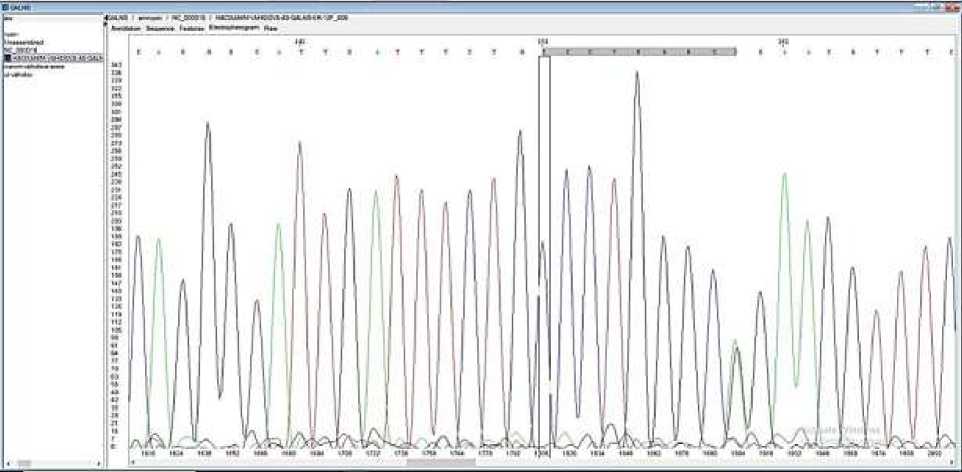
Рисунок 1. Гетерозиготный носитель мутации с.1283 A>G - мать пробанда В.Г.
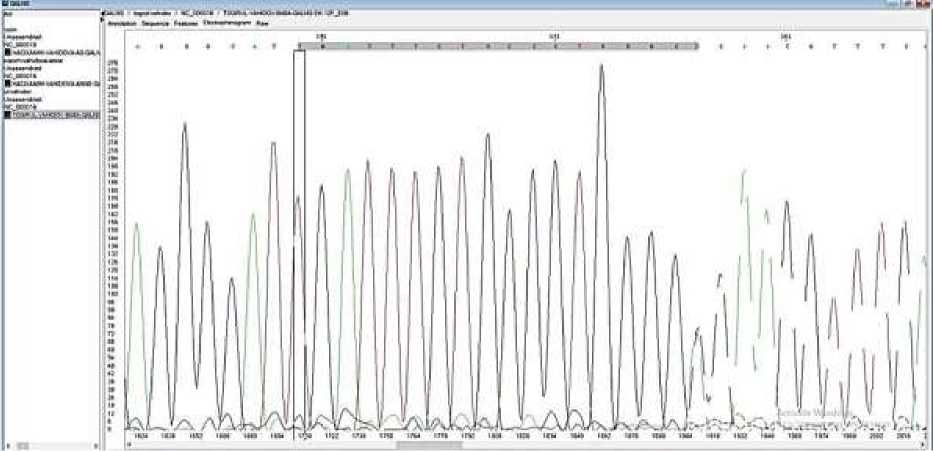
Рисунок.2. Гетерозиготный носитель мутации с.1283 A>G - отец пробанда В.Г.
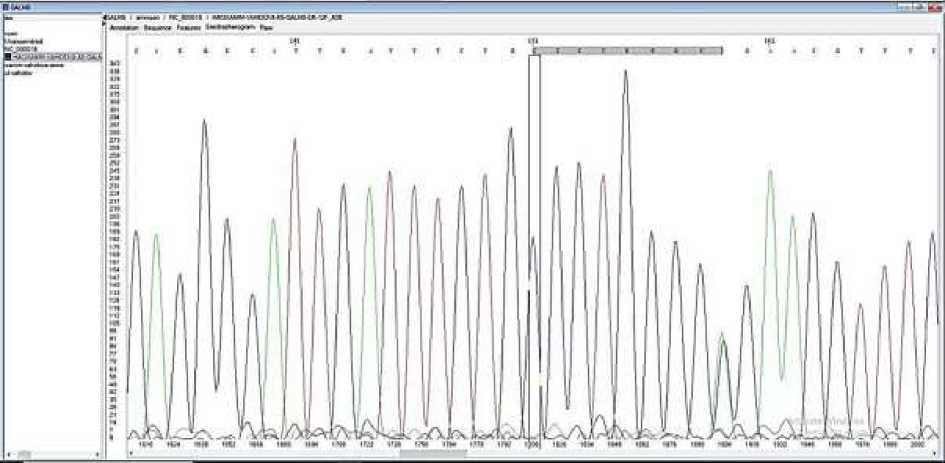
Рисунок 3. Результат пренатальной диагностики - гетерозигоное носительство мутации с.1283 A>G у плода
Пренатальная диагностика плода выявила гетерозиготную мутацию с.1283 A>G гена GALNS, что свидетельствует о здоровом плоде. Родители были ознакомлены с результатами анализа, и по согласию обоих родителей плод был сохранен. Результаты генетического анализа гена GALNS для членов семьи и результат пренатальной диагностики плода представлены в Таблице 3
Таблица 3
РЕЗУЛЬТАТЫ ГЕНЕТИЧЕСКОГО АНАЛИЗА СЕМЬИ ПРОБАНДА В.Г.
|
Пациент |
Замена в гене |
Замена в белке |
Зиготность |
Тип и классификаця |
|
В.Г |
NM_001323544.1:c.1283A>G |
p.(Gln428Arg) |
Гомозигота |
Миссенс-мутация патогенный класс 1 |
|
Мать |
NM_001323544.1:c.1283A>G |
p.(Gln428Arg) |
Гетерозигота |
Миссенс-мутация |
|
Отец |
NM_001323544.1:c.1283A>G |
p.(Gln428Arg) |
Гетерозигота |
Миссенс-мутация |
|
Сестра |
NM_001323544.1:c.1283A>G |
p.(Gln428Arg) |
Гетерозигота |
Миссенс-мутация |
|
ДНК плода |
NM_001323544.1:c.1283A>G |
p.(Gln428Arg) |
Гетерозигота |
Миссенс-мутация |
Таким образом, путем ферментного анализа на наличие всех типов мукополисахаридозов получена низкая активность только для фермента N-ацетил-галактозамин-6-сульфатаза, что свидетельствует о IVA типе болезни. Для уточнения диагноза, установленного ферментным методом, провели дополнительно генетический анализ. Идентифицирована мутация - замена нуклеотида аденин на нуклеотид гуанин в позиции 1283 (A>G) гена GALNS в гомозиготном состоянии. В следствие данной замены происходит замена аминокислоты глицин на аминокислоту аргинин в 428 позиции белка. Мутация 1283 (А>G) является миссенс-мутацией и по классификации относится к патогенному классу 1.
Дополнительное исследование членов семьи для родителей и сестры пробанда показали гетерозиготное состояние выявленной мутации. Пренатальная диагностика плода на 16 неделе беременности выявила гетерозиготную мутацию с.1283 A>G гена GALNS, что подтверждает, что плод здоров. Родители были ознакомлены результатами анализа, и по согласию обоих родителей плод был сохранен для дальнейшего развития.
Выводы
Путем ферментного и генетического анализа удалось выявить гомозиготное состояние мутации с.1283 A>G гена GALNS у 5-летней девочки из Азербайджанской Республики. Обследована вся семья больной девочки. Родители и младшая сестра имели гетерозиготное носительство данной мутации.
Проведена пренатальная диагностика путем амниоцентеза при сроке 16-17 недель беременности, с последующим выделением ДНК из фибробластов и проведением молекулярно-генетических анализов удалось установить гетерозиготное носительство с.1283 A>G у плода.
Планируем проведение пренатальной диагностики всем семьям репродуктивного возраста с наличием больных детей с диагнозом МПС.
Список литературы Первый случай пренатальной диагностики мукополисахаридоза IV типа (синдром Моркио) в Азербайджанской Республике
- Brailsford J. F. Chondro-Osteo-Dystrophy. Roentgenographic and Clinical Features of a Child with Dislocation of Vertebræ // The British Journal of Radiology. 1931. V. 4. №38. P. 83-89. https://doi.org/10.1259/0007-1285-4-38-83
- Tomatsu S., Montaño A. M., Oikawa H., Dung V. C., Hashimoto A., Oguma T., Sly W. S. Enzyme replacement therapy in newborn mucopolysaccharidosis IVA mice: early treatment rescues bone lesions? // Molecular genetics and metabolism. 2015. V. 114. №2. P. 195-202. https://doi.org/10.1016/j.ymgme.2014.05.013
- Borlot F., Arantes P. R., Quaio C. R., Franco J. F. D. S., Lourenço C. M., Gomy I., Kim C. A. Mucopolysaccharidosis type IVA: evidence of primary and secondary central nervous system involvement // American Journal of Medical Genetics Part A. 2014. V. 164. №5. P. 1162-1169. https://doi.org/10.1002/ajmg.a.36424
- Charrow J., Alden T. D., Breathnach C. A. R., Frawley G. P., Hendriksz C. J., Link B., Theroux M. Diagnostic evaluation, monitoring, and perioperative management of spinal cord compression in patients with Morquio syndrome // Molecular Genetics and Metabolism. 2015. V. 114. №1. P. 11-18. https://doi.org/10.1016/j.ymgme.2014.10.010
- Hori T., Tomatsu S., Nakashima Y., Uchiyama A., Fukuda S., Sukegawa K., Orii T. Mucopolysaccharidosis type IVA: common double deletion in the N-acetylgalactosamine-6- sulfatase gene (GALNS) // Genomics. 1995. V. 26. №3. P. 535-542. https://doi.org/10.1016/0888-7543(95)80172-I
- Fukuda S., Tomatsu S., Masuno M., Ogawa T., Yamagishi A., Maruf Rezvi G. M., Orii T. Mucopolysaccharidosis IVA: Submicroscopic deletion of 16q24. 3 and a novel R386C mutation of n‐acetylgalactosamine‐6‐sulfate sulfatase gene in a classical Morquio disease // Human mutation. 1996. V. 7. №2. P. 123-134. https://doi.org/10.1002/(SICI)1098-1004(1996)7:2<123::AIDHUMU6-3.0.CO;2-D
- Jurecka A., Ługowska A., Golda A., Czartoryska B., Tylki-Szymańska, A. Prevalence rates of mucopolysaccharidoses in Poland // Journal of applied genetics. 2015. V. 56. №2. P. 205-210. https://doi.org/10.1007/s13353-014-0262-5
- Biesecker B. B., Peters K. F. Process studies in genetic counseling: peering into the black box // American journal of medical genetics. 2001. V. 106. №3. P. 191-198. https://doi.org/10.1002/ajmg.10004
- Chkioua L., Aloui C., Laradi S., Grissa O., Turkia H. B., Ouesleti S., Froissart R. Genetic heterogeneity of 72 patients with Mucopolysaccharidosis in Tunisia // International Journal of New Technology and Research. 2015. V. 1. №3. P. 263696.
- Masuno M., Tomatsu S., Nakashima Y., Hori T., Fukuda S., Masue M., Orii T. Mucopolysaccharidosis IV A: assignment of the human N-acetylgalactosamine-6-sulfate sulfatase (GALNS) gene to chromosome 16q24 // Genomics. 1993. V. 16. №3. P. 777-778. https://doi.org/10.1006/geno.1993.1266
- Rivera-Colón Y., Schutsky E. K., Kita A. Z., Garman S. C. The structure of human GALNS reveals the molecular basis for mucopolysaccharidosis IV A // Journal of molecular biology. 2012. V. 423. №5. P. 736-751. https://doi.org/10.1016/j.jmb.2012.08.020
- Chuang C. K., Lee C. L., Tu R. Y., Lo Y. T., Sisca F., Chang Y. H., Lin S. P. Nationwide Newborn Screening Program for Mucopolysaccharidoses in Taiwan and an Update of the “Gold Standard” Criteria Required to Make a Confirmatory Diagnosis // Diagnostics. 2021. V. 11. №9. P. 1583. https://doi.org/10.3390/diagnostics11091583
- Alizada S. A., Aliyeva K. A., Musaev Sh. T., Rasulov E. M. Aydin A. S. Genetics of Mucopolysaccharidosis Type IV (Morquio Disorder) in Patients from Azerbaijan // Òîì 7. P. 99.https://doi.org/10.26693/jmbs07.03.099
- Ализаде С.А. Генетика мукополисахаридоза III типа (Синдром Санфилиппо) у детей из Азербайджана // Современная наука: актуальные проблемы теории и практики. Серия: Eстественные и технические науки. 2022. №7/2. С. 5-8. https://doi.org/10.37882/2223-2966.2022.07-2.01
- Wang Z., Zhang W., Wang Y., Meng Y., Su L., Shi H., Huang S. Mucopolysaccharidosis IVA mutations in Chinese patients: 16 novel mutations // Journal of human genetics. 2010. V. 55. №8. P. 534-540. https://doi.org/10.1038/jhg.2010.65
- Nelson J., Crowhurst J., Carey B., Greed L. Incidence of the mucopolysaccharidoses in Western Australia // American journal of medical genetics Part A. 2003. V. 123. №3. P. 310-313. https://doi.org/10.1002/ajmg.a.20314
- Lowry R. B., Applegarth D. A., Toone J. R., MacDonald E., Thunem N. Y. An update on the frequency of mucopolysaccharide syndromes in British Columbia // Human genetics. 1990. V. 85. №3. P. 389-390. https://doi.org/10.1007/BF00206770
- Khan S. A., Peracha H., Ballhausen D., Wiesbauer A., Rohrbach M., Gautschi M., Tomatsu S. Epidemiology of mucopolysaccharidoses // Molecular genetics and metabolism. 2017. V. 121. №3. P. 227-240. https://doi.org/10.1016/j.ymgme.2017.05.016
- Caciotti A., Tonin R., Rigoldi M., Ferri L., Catarzi S., Cavicchi C., Morrone A. Optimizing the Molecular Diagnosis of GALNS: Novel Methods to Define and Characterize Morquio—A Syndrome‐Associated Mutations // Human Mutation. 2015. V. 36. №3. P. 357-368. https://doi.org/10.1002/humu.22751
