Подходы к противоопухолевой терапии на основе модуляции метилирования ДНК

Автор: Максимова В.П., Макусь Ю.В., Попова В.Г., Усалка О.Г., Белицкий Г.А., Якубовская М.Г., Кирсанов К.И.
Журнал: Сибирский онкологический журнал @siboncoj
Рубрика: Обзоры
Статья в выпуске: 4 т.23, 2024 года.
Бесплатный доступ
Актуальность. Метилирование ДНК - важнейший механизм эпигенетической регуляции транскрипции. Нарушения в механизме метилирования ДНК ассоциированы с различными злокачественными новообразованиями (ЗНО), такими как острый миелоидный лейкоз, рак молочной железы, рак предстательной железы и др. Влияние на функциональное состояние ферментов ДНК-метилтрансфераз (DNMTs) и белков семейства ТЕТ (TETs), регулирующих метилирование и деметилирование ДНК, является основой подхода эпигенетической противоопухолевой терапии. В обзоре рассматриваются проблемы и перспективы применения нуклеозидных и ненуклеозидных ингибиторов DNMTs, а также ингибиторов TETs. Представлена оценка результатов клинических исследований эффективности ингибиторов DNMTs, применяемых индивидуально и в составе комбинированной химиотерапии, проведенных за последние 15 лет. Материал и методы. Поиск источников проводили в системах PubMed, ScienceDirect, Web of Science, eLibrary, CyberLeninka. В анализе использовано более 700 публикаций, в обзор включены преимущественно работы последних 10 лет. Ряд статей, опубликованных ранее 2015 г., использован для исторической справки.
Противоопухолевая терапия, метилирование днк, ингибиторы dnmts, ингибиторы tets, клинические испытания
Короткий адрес: https://sciup.org/140307080
IDR: 140307080 | УДК: 616-006-08:577.218 | DOI: 10.21294/1814-4861-2024-23-4-125-140
Approaches to anticancer therapy based on modulation of DNA methylation
Background. DNA methylation is a crucial mechanism of epigenetic regulation of transcription. Disturbances in DNA methylation mechanism are associated with various malignancies such as acute myeloid leukaemia, breast cancer, prostate cancer, etc. Influencing the functional status of DNA methyltransferases (DNMTs) enzymes and TET family proteins (TETs), which regulate DNA methylation and demethylation, is the basis of epigenetic anticancer therapy approach. In this review, we have considered the challenges and prospects of nucleoside and non-nucleoside inhibitors of DNMTs as well as TETs inhibitors. The results of clinical trials on the efficacy of DNMTs inhibitors used individually and as part of combination chemotherapy conducted over the last 15 years are also evaluated. Material and Methods. Sources were searched in PubMed, ScienceDirect, Web of Science, eLibrary, CyberLeninka. More than 700 publications were used in the analysis, but the review included mainly works of the last 10 years. A number of articles published earlier than 2015 were used for historical reference.
Текст обзорной статьи Подходы к противоопухолевой терапии на основе модуляции метилирования ДНК
Общий вклад в развитие и прогрессирование ЗНО вносят генетические нарушения и дисре-гуляция эпигенетических механизмов [1]. Важнейшим механизмом эпигенетической регуляции транскрипции является метилирование ДНК, основанное на добавлении метильного радикала (CH3) к углероду в пятом положении цитозина (5mC) в CpG-динуклеотидах в присутствии S-аденозилметионина (SAM) [2]. Метилирование ДНК катализируется семейством ферментов DNMTs, включающим DNMT1, DNMT3A, DNMT3B, DNMT3L и DNMT3C. Метилирование ДНК подразделяется на поддерживающее метилирование и метилирование de novo. За поддержание установившегося профиля метилирования при делении клетки отвечает метилтрансфераза DNMT1. Метилирование de novo заключается в метилировании прежде не метилированных последовательностей ДНК во время онтогенеза и осуществляется метилтрансферазами DNMT3A и DNMT3B в комплексе с субъединицей DNMT3L, которая повышает каталитическую активность метилтрансфераз, при этом не взаимодействуя с ДНК [3]. Механизмы деметилирования ДНК подразделяются на пассивный и активный. Пассивное деметилирование ДНК происходит в результате дисфункции фермента DNMT1 или при нарушении паттерна метилирования материнской цепи. Активное деметилирование ДНК включает несколько ферментативных реакций, приводящих к удалению или модификации метильной группы. Ферменты семейства Ten-eleven translocation (TET) являются ключевыми участниками в активном деметилировании. Белки TET инициируют окисление 5-метилцитозина до 5-гидроксиметилцитозина (5hmC). Впоследствии TET-опосредованное окисление 5-гидроксиметилцитозина приводит к образованию 5-формилцитозина и 5-карбоксилцитозина [4].
Нарушения в метилировании ДНК приводят к изменению профиля экспрессии генов, хромосомной нестабильности и потере геномного импринтинга, что, в свою очередь, способствует приобретению опухолевыми клетками более агрессивного фенотипа [3]. Аберрантные процессы, связанные с активностью и экспрессией ферментов DNMTs и TETs, характерны для таких ЗНО, как острый миелоидный лейкоз (ОМЛ), рак молочной железы (РМЖ), рак эндометрия, рак предстательной железы (РПЖ) и др. Обратимость эпигенетических модификаций и возможность воздействовать на функционирование эпигенетических ферментов делают белки DNMTs и TETs перспективными мишенями для противоопухолевой терапии [5].
В представленном обзоре приведены данные о терапевтических подходах на основе модуляции метилирования ДНК, рассмотрены уже одобренные или перспективные ингибиторы ферментов DNMTs и TETs, описаны их противоопухолевые эффекты in vitro и in vivo , а также представлены результаты клинических испытаний.
Ингибиторы DNMTs
Ингибиторы DNMTs можно разделить на две группы: нуклеозидные и ненуклеозидные ингибиторы DNMTs.
Нуклеозидные ингибиторы DNMTs
Главной чертой нуклеозидных аналогов цитозина является наличие в их структуре пиримидинового кольца, что обусловливает способность этих молекул встраиваться во вновь синтезированную ДНК и РНК. Азацитидин и децитабин – первые нуклеозидные ингибиторы DNMTs, одобренные Управлением по санитарному надзору за качеством пищевых продуктов и медикаментов США (Food and Drug Administration USA, FDA) для терапии ЗНО. Структурное отличие азацитидина и деци-табина от их природных аналогов, цитидина и 2'-дезоксицитидина, состоит в том, что углерод в положении 5 пиримидинового кольца заменен на атом азота (рис. 1) [6].
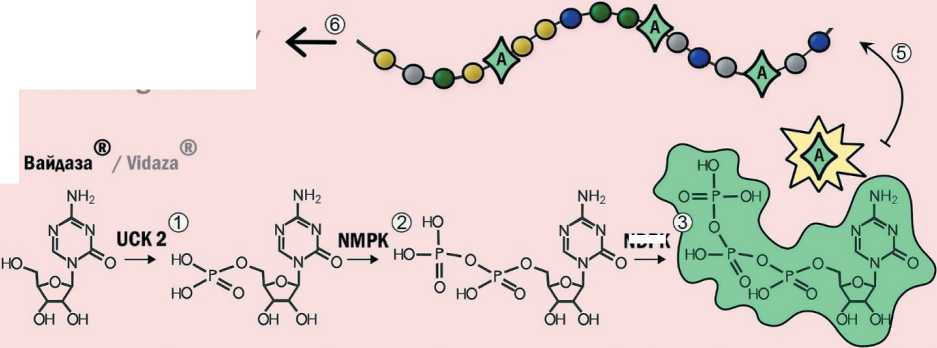
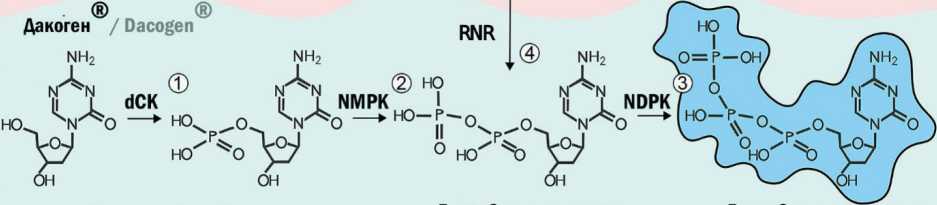
Попадая в клетку, данные соединения подвергаются АТФ-зависимому фосфорилированию и переходят в фармакологически активную форму. На первой стадии азацитидин подвергается действию уридин-цитидинкиназы 2 (UCK2), а децитабин – дезоксицитидинкиназы (dCK), после чего оба агента последовательно фосфорилируются ферментами нуклеозидмонофосфаткиназой (NMPK) и нуклеозиддифосфаткиназой (NDPK), преобразуя азацитидин в 5-азацитидин-5’-трифосфат и деци-табин – в 5-аза-2’-дезоксицитидин-1-5’-трифосфат (рис. 2) [7]. После второго этапа фосфорилирования 5-азацитидин дифосфат под действием рибонуклеотидной редуктазы (RNR) преобразуется в 5-аза-2дезоксицитидин дифосфат.
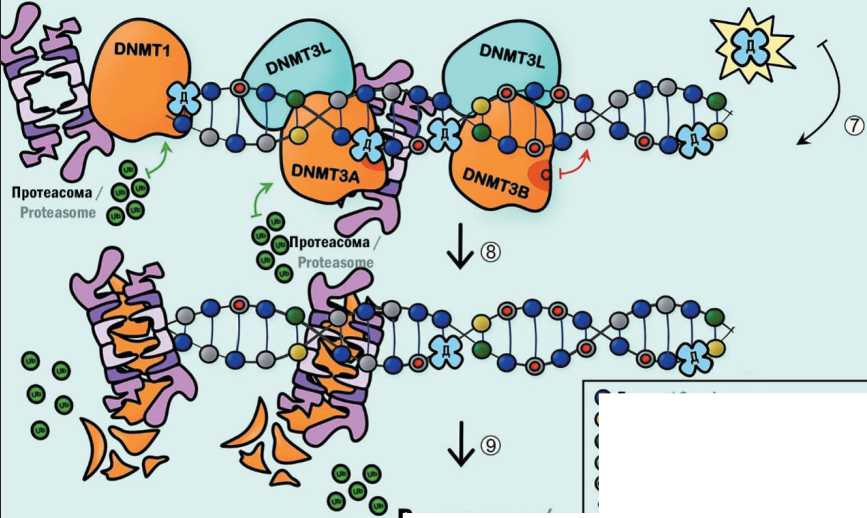
Азацитидин, являясь рибонуклеозидом, включается в последовательность синтезируемой РНК и, в меньшей степени, в ДНК, тогда как децитабин, как аналог дезоксирибозы, включается только в растущие цепи ДНК. После включения в растущую цепь ДНК нуклеозид ковалентно связывается с DNMTs и нарушает сродство ферментов с ДНК, что приводит к деградации комплекса ДНК-DNMTs по убиквитин-зависимому пути и в конечном итоге вызывает гибель клетки [8]. В отсутствие ферментов DNMTs происходит снижение уровня метили-
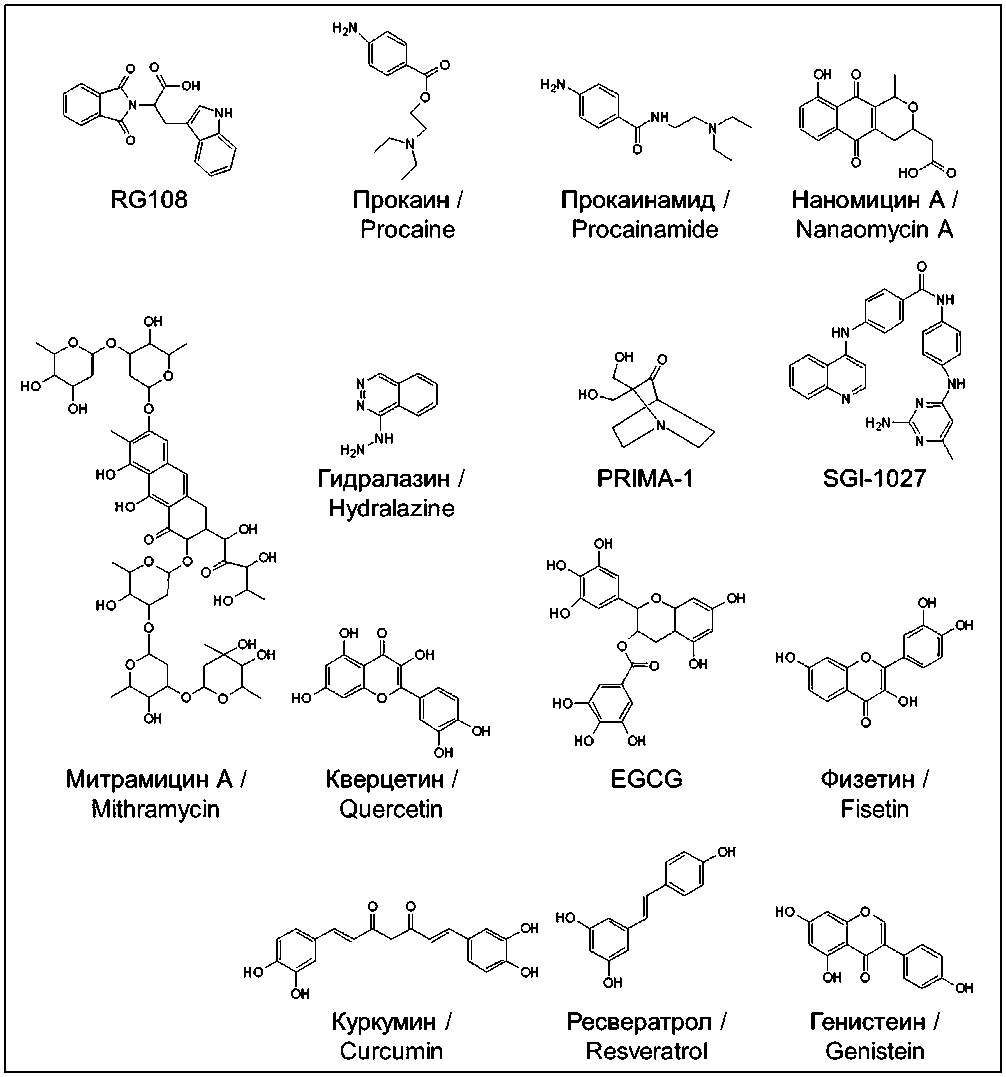
Рис. 1. Химические формулы соединений из группы нуклеозидных ингибиторов DNMT. Примечание: рисунок выполнен авторами
Fig. 1. Chemical formulae of compounds from the group of nucleoside DNMT inhibitors. Note: created by the authors
NDPK
5-азацитидин дифосфат ' 5-azacytidine diphosphate
5-азацитидин трифосфат / 5-azacytidine triphosphate
5-азацитидин монофосфат
5-azacytidine monophosphate
5-азацитидин
5-azacytidine
Деградация РНК/ RNA degradation
5-аза-2-дезоксицитидин / 5-aza-2-deoxycytidine
5-аза-2-дезоксицитидин трифосфат/ 5-aza-2-deoxycytidine triphosphate
5-аза-2-дезоксицитидин дифосфат ' 5-aza-2-deoxycytidine diphosphate
5-аза-2-дезоксицитидин монофосфат
5-aza-2-deoxycytidine monophosphate
Q Гуанин / Guanine
О Аденин /Adenine
Q Тимин/Thymine
О Цитозин/Cytosine
О Метильная группа Methyl group ф 5-азацитидин ' 5-azacitidine ^^5-аза-2-дезоксицитидин / 5-aza-2-de oxycytidine
РепарацияReparation
Рис. 2. Механизмы действия 5-азацитидина и децитабина в клетке:
фосфорилирование азацитидина и децитабина ферментами UCK2 и dCK соответсвенно (1); последовательное фосфорилирование азацитидина и децитабина ферментами NMPK и NDPK с образованием активной формы препарата (2), (3); образование деоксирибонуклеозидной формы азацитидина под действием RNR (4); включение активной формы азацитидина в растущую цепь РНК (5); деградация РНК (6); включение активной формы децитабина в растущую цепь ДНК (7); деградация комплекса ДНК-DNMTпо убиквитин-зависимому пути (8); репарация ДНК(9). Примечание: рисунок выполнен авторами
Fig. 2. Mechanisms of action of 5-azacytidine and decitabine in the cell:
Phosphorylation of azacitidine and decitabine by UCK2 and dCK enzymes respectively (1); Sequential phosphorylation of azacitidine and decitabine by NMPK and NDPK enzymes to form the active form of the drug (2), (3); Formation of deoxyribonucleoside form of azacitidine under the action of RNR (4); Incorporation of the active form of azacitidine into the growing RNA strand (5); RNA degradation (6); Incorporation of the active form of decitabine into the growing DNA strand (7); Degradation of the DNA-DNMT complex via ubiquitindependent pathway (8); DNA repair. Note: created by the authors (9)
рования. В клетке азацитидин и децитабин, являясь аналогами нуклеозидов, подвергаются действию фермента цитидин-дезаминазы, играющей важную роль в метаболизме и утилизации пиримидиновых соединений [9].
Так как первоначально цитотоксичность соединений в отношении опухолевых клеток объяснялась их способностью нарушать синтез ДНК и вызывать ее повреждение, в клинической практике препараты использовали в высоких дозах в качестве антиметаболитов [10]. В 2004 г. азацитидин (Vidaza®, Pharmion Corporation) одобрен для лечения пациентов с миелодиспластическим синдромом (МДС) в качестве гипометилирующего агента [11]. Также азацитидин включен Национальной комплексной онкологической сетью (National Comprehensive Cancer Network, NCCN) и Национальным гематологическим обществом России в рекомендации для терапии ОМЛ [12, 13]. В 2020 г. пероральный азацитидин (Onureg®, Celgene Pty Ltd) одобрен для поддерживающей терапии пациентов с ОМЛ в возрасте 55 лет и старше, находящихся в ремиссии [14]. В 2022 г. FDA одобрило применение азацитидина в комбинации с ивосиденибом (TIB-SOVO®, Servier Pharmaceuticals LLC) для ранее не леченных пациентов с ОМЛ с мутированным геном IDH2 возрастной группы 75 лет и старше или пациентов с сопутствующими заболеваниями, препятствующими проведению интенсивной индукционной химиотерапии (NCT03173248) [15]. Также в 2022 г. азацитидин (Vidaza®, Celgene Corp.) рекомендован FDA для ранее не леченных пациентов с ювенильным миеломоноцитарным лейкозом (NCT02447666) [16]. В 2006 г. децитабин ( Dacogen®, Eisai) одобрен FDA для лечения пациентов с МДС и, так же как и азацитидин, вошел в рекомендации для лечения пациентов с ОМЛ [17]. В 2020 г. FDA одобрило пероральную комбинацию децитабина и цедазуридина (INQOVI®, Astex Pharmaceuticals) для взрослых пациентов с МДС [18]. В настоящее время в России доступно 7 препаратов, в состав которых входит 5-азацитидин: 6 препаратов для подкожного введения (Вайдаза®, Азацитидин-Ника®, Азацитидин-Промомед®, Азацитидин-ТЛ®, Джоцитадин®, Фармазацит®) и 1 препарат в таблетированной форме (Онурег®). Все препараты, за исключением Вайдаза®, применяются для лечения хронического миеломо-ноцитарного лейкоза (ХММЛ) и МДС. Вайдаза® одобрена для терапии хронического миелоидного лейкоза (ХМЛ), ОМЛ и МДС. На рынке представлено 3 препарата с децитабином в качестве активного вещества (Дакоген®, Децитабин Эльфа®, Децитана®), все они показаны при МДС.
В настоящее время проводится более 25 клинических испытаний азацитидина и децитабина для терапии различных ЗНО. Основное направление клинических исследований азацитидина и его комбинаций находится в области гематологических онкозаболеваний. Большое внимание уделяется терапии гемобластозов у пациентов старше 60 лет, а также у пациентов с хроническими заболеваниями, не позволяющими принимать интенсивную химиотерапию. Также активно ведется разработка подходов к терапии солидных опухолей с использованием азацитидина (NCT02260440, NCT00387465, NCT02009436, NCT00748553, NCT03572387). Показано, что использование комбинации азацитидина совместно с венетоклаксом и трансплантацией стволовых клеток приводило к увеличению общей выживаемости пациентов с ОМЛ старше 60 лет [19]. У пациентов с МДС, ХММЛ и ОМЛ было продемонстрировано повышение эффективности терапии азацитидином при совместном режиме с певонедистатом [20]. Для комбинации азацитидина и эназидениба у пациентов с ОМЛ (с мутацией IDH2) было показано двукратное увеличение частоты общего ответа, а также частоты полной ремиссии [21]. Комбинация азацитидина и сорафениба показала высокую эффективность у пожилых пациентов с ранее не леченным ОМЛ (с мутацией FLT3) [22]. Добавление децитабина к режиму химиотерапии, содержащему ритуксимаб, цисплатин, цитарабин и дексаметазон, продемонстрировало эффективность у пациентов с диффузной крупноклеточной В-клеточной лимфомой после неэффективности терапии второй линии [23].
Применение азацитидина и децитабина разрешено в невысоких дозах, позволяющих контролировать побочные эффекты, так как они обладают высоким профилем токсичности и могут приводить к миелосупрессии. Кроме того, данные соединения обладают невысокой стабильностью в водных растворах, а в условиях in vivo подвергаются метаболической деградации при действии цитидиндезаминазы [24]. В связи с этим ведутся активный поиск, разработка и исследование различных аналогов азацитидина и децитабина.
Зебуларин (пиримидин-2-он β-d-рибофуранозид) представляет собой нуклеозидный аналог цитидина, у которого отсутствует аминогруппа в 4-м положении [25]. Помимо репрессии активности DNMTs, зебуларин ингибирует фермент цитидин-деаминазу, что позволяет ему избежать деградации in vivo [26]. Исследования in vitro показали, что зебуларин обладает цитотоксическим эффектом, сопряженным с ингибированием DNMT1, в отношении опухолевых клеток различных нозологий, таких как рак мочевого пузыря, рак толстой кишки и рак поджелудочной железы. При этом зебуларин практически не ингибирует рост нормальных фибробластов человека. Также на различных клеточных линиях было показано, что при действии зебуларина наблюдается повышение уровня экспрессии генов супрессоров опухолей p21 и p16 за счет снижения степени метилирования промоторов этих генов [27, 28]. На клетках MCF7 и MDA-MB-
231 in vitro было показано, что вызванное зебула-рином ингибирование белков DNMT1, DNMT3A и DNMT3B, а также белков MBD2 и MBD3 приводит к реактивации экспрессии гена ESR1 , что может повысить чувствительность клеток к гормональной противоопухолевой терапии. Также зебуларин в комбинации с децитабином и вориностатом проявляет синергическое противоопухолевое действие и реактивирует экспрессию эпигенетически репрессированных генов супрессоров опухолей в клетках РМЖ MCF7, MDA-MB-231 и клетках рака мочевого пузыря T24 [29]. Исследование in vivo на модели лейкоза мышей L1210 показало, что добавление зебуларина к терапии децитабином вызывает увеличение содержания децитабина в плазме крови и, как следствие, увеличение эффективности терапии [30]. Исследования фармакодинамики на мышах, собаках и обезьянах показали низкую биодоступность, быстрый период полувыведения вещества и обратимость ингибирования DNMTs зебуларином. Опубликованные данные свидетельствуют об ограниченном потенциале зебуларина в клинических исследованиях [31].
FdCyd (5-Фтор-2’-дезоксицитидин) представляет собой аналог цитидина, у которого атом водорода в положении C5 замещен на атом фтора. После встраивания в ДНК FdCyd ковалентно связывается с DNMTs и оказывает ингибирующее действие на стадии β-элиминирования реакции переноса метильной группы из-за низкой реакционной способности, поскольку атом фтора является плохо уходящей группой. FdCyd стабилен в водных растворах, а также менее токсичен, чем 5-азацитидин и 5-аза-2’-дезоксицитидин. Однако в условиях in vivo он метаболизируется цитидиндезаминазой в фармакологически активные токсичные метаболиты 5-фтор-2'-дезоксиуридин, 5-фторурацил и 5-фторуридин, что может быть препятствием для применения FdCyd исключительно в качестве деметилирующего агента [32]. Для повышения стабильности соединения in vivo было предложено использовать его в комбинации с ингибитором цитидиндеаминазы – тетрагидроуридином (THU). Результаты исследований свидетельствуют, что оба соединения стабильны в растворе [33], а присутствие THU увеличивает содержание FdCyd в 4 раза и биодоступность до 24 % [34]. В настоящее время проходит ряд клинических испытаний FdCyd в отношении солидных опухолей, таких как РМЖ, немелкоклеточный рак легкого и ЗНО головы или шеи [35].
CP-4200 (5-азацитидин-5'-элайдат) является производным 5-азацитидина и элаидиновой кислоты. Наличие эфирной группы обеспечивает поглощение соединения по пути, не зависимому от транспортных систем нуклеозидов [36]. M. Rius et al. [37] показано in vitro, что транспортные белки hCNT1, hCNT3 и hENT1 регулируют поглощение азацитидина, в то время как для CP-4200 наблюдается низкая аффинность к данным белкам. Деметилирующие свойства CP-4200 сравнимы со свойствами азацитидина. CP-4200 ингибирует активность DNMT1, что сопровождается как глобальным деметилированием ДНК на уровне всего генома, так и деметилированием промоторов генов супрессоров опухолевого роста (например, TIMP-3, DAPK-1), что приводит к устойчивой активации их экспрессии [36]. В настоящее время продолжаются доклинические исследования агента.
NPEOC-DAC (2'-Дезокси-N4-(2-(4-нитрофенил) этоксикарбонил)-5-азацитидин) представляет собой 5-аза-2’-деоксицитидин с модификацией положения N4 азацитидинового кольца. В условиях in vivo при действии фермента карбоксилэстеразы 1 (CES1) происходит расщепление эфирной связи в молекуле NPEOC-DAC, что приводит к высвобождению децитабина. Однако исследования in vitro на клетках РМЖ MCF7, рака мочевого пузыря T24 и рака печени Hep G2 продемонстрировали, что NPEOC-DAC в 23 раза менее эффективен, чем децитабин [38]. Кроме того, к недостаткам данной молекулы относятся гепатотоксичность и возможность применения только при опухолях с высокой экспрессией карбоксилэстераз. Исследований противоопухолевой активности NPEOC-DAC in vivo в литературе не представлено.
Гвадецитабин (SGI-110) представляет собой молекулу 5-аза-2'-дезоксицитидина, модифицированную дезоксигуанозином, который обеспечивает устойчивость соединения к действию фермента цитидиндезаминазы. SGI-110 индуцировал гипометилирование промоторов генов семейства CTA в клеточных линиях лейкоза HL60 и KG1 [39]. В литературе представлены результаты клинических испытаний гвадецитабина (фазы II-III) в терапии как гематологических ЗНО (NCT02348489, NCT02907359, NCT02920008), так и солидных опухолей (ISRCTN 16332228, NCT01896856, NCT02901899, NCT01696032). Гвадецитабин обладает близкой с децитабином противоопухолевой эффективностью в отношении МДС, но менее эффективен при ОМЛ. При этом наблюдалась связь между терапевтическим ответом и степенью деметилирования ДНК пациентов [40]. Комбинация гвадецитабина с карбоплатином у пациенток с распространенным раком яичника показала двукратное увеличение бессобытийной выживаемости и частоту общего ответа по сравнению со стандартной терапией [41]. У пациенток с платино-резистентным раком яичника показатель клинической пользы комбинации гвадецитабина с пембролизумабом составил 31,4 % [42]. В отношении солидных опухолей добавление гвадецитабина к комбинации цисплатина с гемцитабином приводило к практически двукратному увеличению выживаемости без прогрессирования относительно контрольной группы [43].
Ненуклеозидные ингибиторы DNMTs
Отличительной особенностью ненуклеозидных соединений является способность взаимодействовать с каталитическим центром ферментов DNMTs [44]. Механизмы действия большинства соединений изучены недостаточно, однако общим для данных агентов является их способность снижать уровень метилирования генов супрессоров опухолей и реактивировать их транскрипцию (рис. 3).
RG108 (N-Фталил-L-триптофан) – производное триптофана, действие которого основано на инактивации фермента DNMT1 за счет взаимодействия с активным центром фермента. RG108 является первым таргетным ненуклеозидным ингибитором DNMTs [45]. На клеточной линии рака эндометрия Ishikawa in vitro показано, что RG108 ингибирует активность DNMT3B. При действии RG108 происходит деметилирование промотора гена MLH1 [46]. В исследовании in vivo на ксенографтной модели рака пищевода Eca-109 продемонстрировано, что RG-108 способствует повышению чувствительности клеток рака пищевода к радиотерапии, которое сопровождается глобальным изменением экспрессии генов различных сигнальных путей [45]. Кроме того, на мезенхимальных стволовых клетках из костного мозга свиньи показано, что RG108 способен реактивировать экспрессию генов плюрипотентности NANOG и POU5F1 . Клинические испытания RG108 пока не зарегистрированы.
Прокаин и прокаинамид представляют собой производные 4-аминобензойной кислоты, которые одобрены FDA для применения в качестве анестетика и антиаритмического препарата соответственно. Данные соединения ингибируют активность метилтрансферазы DNMT1 за счет снижения сродства фермента как к ДНК, так и к S-аденозил-
Наномицин А и Митрамицин А – антибиотики, выделенные из актинобактерий рода Streptomyces. Наномицин А используется в качестве противо-
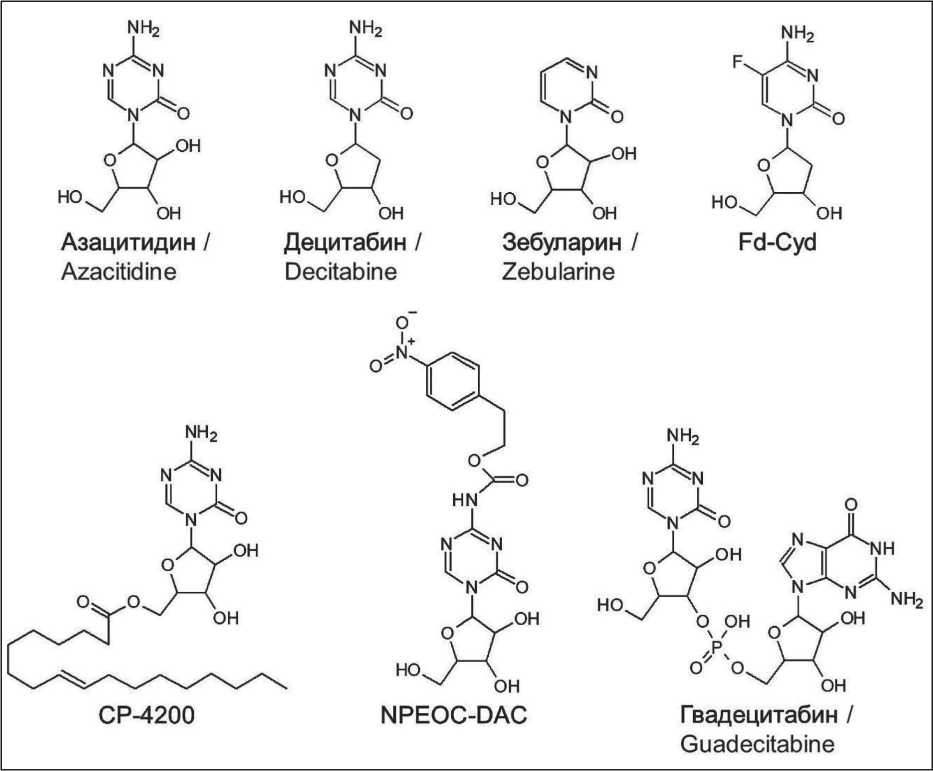
Рис. 3. Химические формулы соединений из группы ненуклеозидных ингибиторов DNMTs. Примечание: рисунок выполнен авторами
Fig. 3. Chemical formulae of compounds from the group of non-nucleoside inhibitors of DNMTs. Note: created by the authors
грибкового препарата в ветеринарной практике, а митрамицин А является противоопухолевым антибиотиком, который ранее применяли для терапии нескольких типов ЗНО, включая ХМЛ, ОМЛ, рак яичка [55, 56]. Наномицин А селективно ингибирует DNMT3B путем взаимодействия с каталитическим центром фермента, что приводит к снижению уровня метилирования в клетках рака легких A549, колоректального рака HCT116 и лейкоза HL60. Также в клетках рака легких A549 наблюдалась реэкспрессия гена RASSF1 [57]. В то же время наномицин вызывал активацию онкогенов HMOX1 , NQO1 , AKR1C1 , AKR1C2 , GCLM и PPT2 в клетках нейробластомы BE(2)-c и CHP134 [58]. Митрамицин А связывается с CG-богатыми последовательностями, что приводит к стерическому ингибированию метилирования ДНК, а также к нарушению взаимодействия транскрипционных факторов с ДНК. С помощью молекулярного докинга также показана способность антибиотика взаимодействовать с каталитическим центром DNMTs. Исследования in vitro продемонстрировали, что под действием митрацимицина А в клетках рака легких CL1-5 происходит реактивация эпигенетически репрессированных генов SLIT2 и TIMP-3 [59]. Из-за токсичности применение ми-трамицина ограничено, однако ведется активная разработка его производных.
Гидралазин (1(2Н)-фталазинонгидразон) – вазодилатирующее гипотензивное средство, используемое для лечения артериальной гипертензии и сердечной недостаточности [60]. На клетках РПЖ Du-145 и PC3 было показано, что гидралазин снижает экспрессию генов DNMT1 , DNMT3A и DNMT3B , а также уровень фермента DNMT1 [61]. Наиболее вероятным механизмом действия гидралазина является его прямое взаимодействие с ферментом DNMT1 [62]. Для гидралазина показан противоопухолевый эффект на различных клеточных моделях [61, 63]. Зарегистрировано несколько клинических исследований (фазы I–III) эффективности гидралазина в комбинации с ингибитором HDAC вальпроатом для терапии ХМЛ, кожной Т-клеточной лимфомы и солидных опухолей (NCT0096060, TPVGH 97-07-07). Комбинация показала негативные результаты для пациентов с солидными опухолями, при этом для пациентов с ХМЛ и кожной Т-клеточной лимфомой медиана выживаемости без прогрессирования заболевания составила более 36 и 18 мес соответственно [64, 65].
PRIMA-1 и его метилированная форма PRIMA-1Met (APR246, эпренетапопт) – низкомолекулярные соединения, влияющие на конформацию мутантного белка p53, что приводит к восстановлению его способности активировать экспрессию респонсив-ных генов [66]. В 2014 г. показано, что PRIMA-1 индуцирует глобальное деметилирование ДНК и увеличивает степень гидроксиметилирования ДНК в клетках рака щитовидной железы с мутантным p53. Наблюдаемые эффекты обусловлены снижением уровня мРНК генов DNMT1, DNMT3A и DNMT3B и повышением экспрессии генов ТЕТ1 и ТЕТ2 [67]. Кроме того, PRIMA-1 вызывает деметилирование гена TP73 в клетках множественной миеломы [68]. При этом данных о деметилирующих способностях APR246 в литературе не представлено. Противоопухолевые свойства PRIMA-1 и APR246 были продемонстрированы in vitro и in vivo для таких ЗНО, как острый промиелоци-тарный лейкоз, крупная B-клеточная лимфома, меланома, множественная миелома, РПЖ, РМЖ и др. [69–71]. Фаза II клинических исследований продемонстрировала синергетическое действие комбинации APR246 с 5-азацитидином при терапии пациентов с ОМЛ и МДС, частота общего ответа составила 71 и 73 % соответственно. В апреле 2019 г. FDA предоставило APR-246 статус Fast Track и OrphanDrug для лечения МДС у взрослых с мутацией TP53 [72].
SGI-1027 (N-(4-(2-амино-6-метилпиримидин-4-иламино)фенил)-4-(хинолин-4-иламино)бензамид) является представителем нового класса низкомолекулярных ингибиторов ферментов DNMTs на основе липофилина [73]. Механизм действия агента заключается в конкуренции с донором метильной группы SAM за доступ к сайту связывания фермента. Было показано, что SGI-1027 способен ингибировать DNMT1 в клетках ХМЛ [74]. Несмотря на то, что SGI-1027 оказывает минимальный токсический эффект на опухолевые клетки, соединение может стать перспективным кандидатом для использования в комбинированной противоопухолевой терапии.
Кверцетин (3,3`,4`,5,7-пентагидроксифлава-нон) – соединение растительного происхождения из класса флавоноидов, содержащееся в различных фруктах, овощах, зеленом чае и листьях лекарственных растений [75]. Данный флавоноид используется в качестве антиоксидантного и терапевтического агента в аллергологии, урологии и т.д. [76]. В исследовании in vivo с ксенограф-тами острого промиелоцитарного лейкоза HL-60 при действии кверцетина показано значительное снижение экспрессии белков DNMT1 и DNMT3A [77]. С помощью молекулярного докинга показано, что кверцетин способен связываться с активными центрами белков DNMTs и ингибировать их активность. Также продемонстрировано, что кверцетин конкурентно связывается с метилтранферазами EZH2 и G9a в месте взаимодействия белков с DNMTs. На клетках рака шейки матки HeLa TI показано дозозависимое снижение экспрессии генов DNMTs при действии кверцетина, также наблюдалось снижение уровня метилирования промоторов генов супрессоров опухолевого роста APC, CDH1, CDH13, DAPK1, FHIT, GSTP1, MGMT, MLH1, PTEN, RARB, RASSF1, SOC51, TIMP3 и VHL [78], также в исследовании in vitro на клетках острого промиелоцитарного лейкоза HL-60 человека кверцетин проявил деметилирующий эффект в промоторах генов BCL2L11 и DAPK1. Планируется фаза II клинического испытания кверцетина в качестве препарата для лечения пациентов с плоскоклеточной карциномой (NCT03476330) и раком ротовой полости (NCT05456022).
EGCG ((-)-эпигаллакатахин-3-галлат) – катехин зеленого чая, обладающий многообразием свойств, в том числе противоопухолевыми и антиканцерогенными [79]. Противоопухолевый эффект соединения показан в исследованиях in vitro и in vivo на моделях РМЖ, рака пищевода, рака мочевого пузыря, немелкоклеточного рака легкого и лейкозов [80, 81]. На различных моделях in vitro показано, что при действии EGCG происходит значительное снижение экспрессии генов DNMT1 , DNMT3A и DNMT3B, сопровождаемое активацией транскрипции генов супрессоров опухолей. В клетках РМЖ MDA-MB-231 и MCF7 наблюдалась реактивация экспрессии гена SCUBE2 [82], в клетках рака шейки матки HeLa – генов RARβ , CDH1 и DAPK1 [83], в клетках меланомы A431 – генов CDKN1A и CDKN2A [84], в клетках ОМЛ U-937 – гена SOCS1 [85], в клетках РМЖ MCF7 – гена IFI16 [86]. На клетках рака пищевода KYSE 150 показано, что EGCG способен вызывать реактивацию генов MGMT , MLH1 , WIF-1 , TERT [87]. На клеточной линии плоскоклеточной карциномы полости рта CAL-27 в ходе анализа метилома выявлено, что EGCG вызывает дифференциальное метилирование более 700 генов, относящихся к таким процессам, как клеточный транспорт, клеточный цикл, окислительные процессы и апоптоз [88]. В исследовании in vitro на клетках РМЖ MCF7 продемонстрировано, что EGCG индуцирует экспрессию гена IFI16 , который ассоциирован с интерфероновым сигналингом и запуском врожденного клеточного иммунитета в опухолевых клетках [86]. Клинических испытаний EGCG для терапии ЗНО не зарегистрировано, однако есть несколько исследований зеленого чая в качестве сопутствующей терапии при РМЖ и РПЖ. У пациентов с РПЖ показано, что EGCG снижает уровни фактора роста гепатоцитов (HGF) и фактора роста эндотелия сосудов (VEGF) [89].
Физетин (3,3',4',7-тетрагидроксифлавон) – растительный флавоноид из группы полифенолов, содержится в овощах, фруктах и ягодах. Физетин обладает широким спектром действия, проявляя противоаллергическую, противоопухолевую и нейропротекторную активность [90]. Показана способность физетина слабо ингибировать DNMT SssI в бактериях E. coli и DNMT1 в клетках РМЖ. В исследованиях in vitro на клетках рака поджелудочной железы PANC-1 и BxPC-3, а также in vivo с ксенографтами этих же клеточных линий продемонстрировано дозозависимое снижение жизнеспособности клеток при действии физети- на. При этом обнаружено, что физетин вызывает активацию аутофагии в клетках рака поджелудочной железы [91]. Физетин способен связываться с β-тубулином и стабилизировать микротрубочки с большей эффективностью, чем паклитаксел, что было показано на клетках РПЖ PC-3 и Du-145. Кроме того, при действии физетина в клетках рака яичников NCI/ADR-RES происходит подавление экспрессии Р-гликопротеина, гиперэкспрессия которого ассоциирована с развитием множественной лекарственной устойчивости [92]. Данных о клинических испытаниях физетина в литературе не представлено.
Куркумин (диферулоилметан) – полифеноль-ное соединение с противовоспалительным, антиоксидантным и противоопухолевым действиями. Молекулярная активность куркумина включает в себя ингибирование протеасом, ингибирование тирозинкиназ, а также инактивацию различных онкогенов [93]. Куркумин блокирует каталитический сайт C1226 фермента DNMT1, что обусловливает его гипометилирующее действие [94]. На клетках ОМЛ THP-1, Kasumi-1, MV4-11 и ML-1 показано, что куркумин вызывает ингибирование экспрессии DNMT1 как на уровне мРНК, так и на уровне белка. При этом агент не влиял на экспрессию DNMT1 в моноцитах периферической крови. При действии куркумина отмечено деметилирование промотора гена супрессора опухолей CDKN2B , обусловливающее его реэкспрессию [95]. В то же время на клетках множественной миеломы продемонстрирована способность куркумина репрессировать транскрипцию онкогена MTOR посредством гиперметилирования промотора. Подобный эффект объясняется гиперактивностью ферментов DN-MT3A и DNMT3B, вызванной куркумином [96]. На модели РМЖ in vitro выявлено, что куркумин вызывает деметилирование промотора и реактивацию экспрессии гена BRCA1 [97]. На клетках колоректального рака HCT116, HT29 и RKO показано, что куркумин деметилирует ДНК более чем в тысяче CpG-локусов [98]. В недавнем исследовании на клетках колоректального рака HCT116 продемонстрировано, что куркумин в комбинации с 5-азацитидином могут быть эффективной альтернативой в терапии опухолей, резистентных к децитабину [99]. В настоящее время куркумин проходит клинические испытания для терапии РМЖ, колоректального рака, рака поджелудочной железы и РПЖ (NCT01490996). Добавление куркумина к режиму химиотерапии FOLFOX при метастатическом колоректальном раке привело к увеличению общей выживаемости более чем в 2 раза [100]. У больных РМЖ добавление куркумина к терапии паклитакселом способствовало повышению частоты общего ответа до 50,7 % vs 33 % в контрольной группе [101].
Ресвератрол (3,5,4'-тригидроксистильбен) – полифенол природного происхождения, фитоалексин, содержится в корнях морозника белого Veratrum grandi^orum O. Loes, а также в кожуре фруктов, какао и орехах. Ресвератрол имеет широкий спектр биологических свойств, включая антигликирующее, антиоксидантное, противовоспалительное, нейропротекторное, противоопухолевое и антивозрастное действие [102]. С помощью молекулярного докинга и сцинтилляционного бесконтактного анализа производных ресвератрола получены данные о взаимодействии лигандов с каталитическими центрами белков DNMT3A и DNMT3B, но не DNMT1 [103]. В клетках РМЖ MCF7 ресвератрол вызывает значительное снижение активности DNMTs и уровней экспрессии, связывающих метил-CpG белков MeCP2 и MBD2, что коррелирует с понижением жизнеспособности клеток [104]. Результаты клинических испытаний ресвератрола свидетельствуют о его невысокой эффективности при терапии ЗНО.
Генистеин (4',5,7-тригидроксиизофлавон) – фитонутриент из класса изофлавонов, является природным фитоэстрогеном, в большом количестве содержится в бобовых, семенах, фруктах и овощах. Генистеин обладает антиоксидантными, противовоспалительными и антибактериальными свойствами, в частности, его применяют для облегчения симптомов менопаузы, профилактики сердечнососудистых заболеваний, остеопороза, а также для снижения заболеваемости некоторыми видами рака [105]. С помощью молекулярного докинга показана способность генистеина взаимодействовать с каталитическим центром ферментов DNMT1, DNMT3A и DNMT3B. На клетках HeLa и MCF7 генистеин вызывал времязависимое снижение экспрессии DNMT1, DNMT3A и DNMT3B как на уровне мРНК, так и на уровне белка [106]. Подавление экспрессии DNMT3A и DNMT3B было продемонстрировано и для комбинации генистеина с сульфорафаном и бутиратом натрия на клетках РМЖ MDA-MB-231 и MCF7 [107]. Также на клетках РМЖ MCF7 и MDA-MB-231 показано снижение глобального уровня метилирования ДНК, в том числе в промоторных областях таких генов супрессоров опухолевого роста, как ATM, APC, PTEN, SERPINB5, BRCA1 [108, 109]. На клетках РПЖ Du-145 и LNCaP продемонстрировано, что комбинация генистеина и фитоэстрогена дайдзеина влияет на профиль метилирования 58 генов, включая MAD1L1, TRAF7, KDM4B и hTERT. На ксенографтах нейробластомы SK-N-SH показано, что при действии генистеина происходят реактивация транскрипции генов супрессоров опухолей TP53 и CHD5 и снижение экспрессии DNMT3B [110]. Фазы I–II клинических испытаний генистеина для терапии РПЖ, колоректального рака и др. показали, что он хорошо переносится и не приводит к увеличению числа побочных эффектов [111]. Однако до сих пор не было получено весомых доказательств эффективности генистеина в терапии ЗНО. В настоящее время генистеин исследуется как часть диеты пациентов с раком поджелудочной железы и раком мочевого пузыря для снижения побочных эффектов химиотерапии (NCT02336087, NCT01489813).
Ингибиторы ТЕТ
Разработка ингибиторов метилцитозиновых диоксигеназ ТЕТ началась в 2018 г. с молекул Bobcat212 и Bobcat339 [112]. В исследованиях in vitro продемонстрирована их способность ингибировать гидрокисметилирование ДНК, что опосредовано воздействием на белки TETs [112]. Однако в 2022 г. опубликовано опровержение механизма действия Bobcat339 в отношении TETs. Исследователями во главе с Н. Третьяковой с помощью анализа LC-ESI-MS показано, что Bobcat339 обладает минимальной ингибирующей активностью в отношении TET1 и TET2 человека. Основной причиной ложноположительного результата по ингибированию ферментов было наличие в растворе ионов меди (II), а не активность соединения [113]. В 2020 г. с помощью виртуального скрининга лигандов (Lvspipe) и библиотеки соединений Национального института рака (National Cancer Institute, NCI) выявлен первый ингибитор ТЕТ – низкомолекулярное соединение C35, которое специфически блокирует каталитическую активность метилцитозиновых диоксигеназ. Молекулярное моделирование показало, что C35 может частично занимать сайт связывания ДНК с субстратом и блокировать каталитический центр TET2, отталкивая Fe (II), что в результате приводит к истощению метки 5-hmC [114]. TETi76 синтезирован на основе каркаса 2HG в αKG-связывающем сайте каталитического домена TET2. Показано, что 2-метил- и 4-4-гидроксипроизводное αKG в составе TETi76 имеют решающее значение для поддержания ингибирующей активности. В исследовании in vitro показано, что TETi76 индуцирует апоптотическую гибель клеток лейкоза время- и дозозависимым образом. Также in vivo продемонстрировано, что TETi76 значительно снижает рост опухолевых клеток ксенотрансплантатов ОМЛ человека с мутацией TET2 [115].
Заключение
Злокачественные новообразования характеризуются глобальной перестройкой эпигенетической системы регуляции транскрипции. Ключевым механизмом данной системы является метилирование ДНК. Нарушения в механизме метилирования ДНК приводят к изменению хроматинового ландшафта и, как следствие, изменению паттерна экспрессии генов, вовлеченных в ключевые процессы клеточной регуляции. Основными причинами аберрантного метилирования ДНК являются делеции, мутации или нарушение активности регуляторных ферментов DNMTs и ТЕТs. Для некоторых нозологий DNMTs и TETs в приобретении опухолью агрессивного фенотипа играют значительную роль, что может быть использовано в противоопухолевой терапии.
В настоящее время терапия ЗНО на основе модуляции метилирования ДНК включает в себя использование только двух нуклеозидных ингибиторов DNMTs – азацитидина и децитабина. Данные агенты эффективно применяются в терапии МДС и ОМЛ как в России, так и за рубежом. Однако при длительном приеме может развиваться резистентность к 5-азацитидину и децитабину, что осложняет дальнейшее лечение. Использование ингибиторов DNMTs из других химических классов, а также селективных ингибиторов TETs могло бы способствовать решению этой проблемы. Другой проблемой при использовании эпигенетических препаратов является их высокая токсичность. Несмотря на то, что деметилирующее действие агентов не вносит значимых изменений на уровне генома, а уровень метилирования ДНК восстанавливается до исходных значений уже через 14 дней после окончания приема препаратов, профиль токсичности азацитидина и децитабина соответствует другим аналогам нуклеозидов и алкилирующим агентам, взаимодействующим с ДНК. Это актуализирует поиск новых эффективных соединений-деметиляторов с меньшей токсичностью.
Ранее одной из главных проблем эпигенетической терапии была низкая эффективность эпигенетических модуляторов в отношении солидных опухолей. При индивидуальном действии ингибиторы DNMTs действительно проявляют ограничен- ную эффективность в клинических исследованиях. Однако разнообразие молекулярных эффектов деметилирующих агентов позволяет использовать их в комбинации с уже применяемыми препаратами для повышения эффективности противоопухолевой терапии. Такой подход привел к значительному прогрессу в этом направлении. За последние 5 лет получен ряд положительных результатов клинических испытаний применения как нуклеозидных, так и ненуклеозидных ингибиторов DNMTs в комбинированной терапии. Из группы нуклеозидных ингибиторов DNMTs к наиболее перспективным агентам для комбинированной терапии можно отнести децитабин и гвадецитабин. Для этих препаратов по результатам клинических испытаний показаны следующие эффективные сочетания: децитабин и ритуксимаб для терапии диффузной В-крупноклеточной лимфомы; гвадецитабин и иринотекан для терапии колоректального рака; гвадецитабин и карбоплатин для терапии рака яичников. Из группы ненуклеозидных ингибиторов DNMTs наибольшую эффективность показал куркумин: в комбинации с линией FOLFOX для терапии колоректального рака и в комбинации с паклитакселом для терапии РМЖ. Повышение эффективности комбинированной терапии опухолей с аберрантным эпигенетическим профилем может быть достигнуто за счет разработки протоколов прецизионной медицины на основе высокотехнологичных методов анализа профиля мутаций и экспрессии ферментов DNMTs и TETs в сочетании с современными клиническими протоколами.
Список литературы Подходы к противоопухолевой терапии на основе модуляции метилирования ДНК
- Hanahan D. Hallmarks of Cancer: New Dimensions. Cancer Discov. 2022; 12(1): 31-46. https://doi.org/10.1158/2159-8290.CD-21-1059.
- Cheng Y., He C., Wang M., Ma X., Mo F., Yang S., Han J., Wei X. Targeting epigenetic regulators for cancer therapy: mechanisms and advances in clinical trials. Signal Transduct Target Ther. 2019; 4: 62. https://doi.org/10.1038/s41392-019-0095-0.
- Maksimova V.P., Usalka O.G., Makus' Yu.V., Popova V.G., Trapeznikova E.S., Khairieva G.I., Sagitova G.R., Zhidkova E.M., Prus A.Yu., Yakubovskaya M.G., Kirsanov K.I. Narushenie metilirovaniya DNK pri zlokachestvennykh novoobrazovaniyakh. Uspekhi molekulyarnoi onkologii. 2022; 9(4): 24-40. https://doi.org/10.17650/2313-805X-2022-9-4-24-40.
- Kohli R.M., Zhang Y. TET enzymes, TDG and the dynamics of DNA demethylation. Nature. 2013; 502(7472): 472-9. https://doi.org/10.1038/nature12750.
- Takeshima H., Niwa T., Yamashita S., Takamura-Enya T., Iida N., Wakabayashi M., Nanjo S., Abe M., Sugiyama T., Kim Y.J., Ushijima T. TET repression and increased DNMT activity synergistically induce aberrant DNA methylation. J Clin Invest. 2020; 130(10): 5370-9. https://doi.org/10.1172/JCI124070.
- Liu X.L., Liu H.Q., Li J., Mao C.Y., He J.T., Zhao X. Role of epigenetic in leukemia: From mechanism to therapy. Chem Biol Interact. 2020; 317. https://doi.org/10.1016/j.cbi.2020.108963.
- Stresemann C., Lyko F. Modes of action of the DNA methyltransferase inhibitors azacytidine and decitabine. Int J Cancer. 2008; 123(1): 8-13. https://doi.org/10.1002/ijc.23607.
- Yang X., Lay F., Han H., Jones P.A. Targeting DNA methylation for epigenetic therapy. Trends Pharmacol Sci. 2010; 31(11): 536-46. https://doi.org/10.1016/j.tips.2010.08.001.
- Gu X., Tohme R., Tomlinson B., Sakre N., Hasipek M., Durkin L., Schuerger C., Grabowski D., Zidan A.M., Radivoyevitch T., Hong C., Carraway H., Hamilton B., Sobecks R., Patel B., Jha B.K., Hsi E.D., Maciejewski J., Saunthararajah Y. Decitabine- and 5-azacytidine resistance emerges from adaptive responses of the pyrimidine metabolism network. Leukemia. 2021; 35(4): 1023-36. https://doi.org/10.1038/s41375-020-1003-x.
- Malik P., Cashen A.F. Decitabine in the treatment of acute myeloid leukemia in elderly patients. Cancer Manag Res. 2014; 6: 53-61. https://doi.org/10.2147/CMAR.S40600.
- Kaminskas E., Farrell A.T., Wang Y.C., Sridhara R., Pazdur R. FDA drug approval summary: azacitidine (5-azacytidine, Vidaza) for injectable suspension. Oncologist. 2005; 10(3): 176-82. https://doi.org/10.1634/theoncologist.10-3-176.
- Tallman M.S., Wang E.S., Altman J.K., Appelbaum F.R., Bhatt V.R., Bixby D., Coutre S.E., De Lima M., Fathi A.T., Fiorella M., Foran J.M., Hall A.C., Jacoby M., Lancet J., LeBlanc T.W., Mannis G., Marcucci G., Martin M.G., Mims A., O'Donnell M.R., Olin R., Peker D., Perl A., Pollyea D.A., Pratz K., Prebet T., Ravandi F., Shami P.J., Stone R.M., Strickland S.A., Wieduwilt M., Gregory K.M.; OCN; Hammond L., Ogba N. Acute Myeloid Leukemia, Version 3.2019, NCCN Clinical Practice Guidelines in Oncology. J Natl Compr Canc Netw. 2019; 17(6): 721-49. https://doi.org/10.6004/jnccn.2019.0028.
- Savchenko V.G., Parovichnikova E.N., Afanas'ev B.V., Gritsaev S.V., Semochkin S.V., Bondarenko S.N., Troitskaya V.V., Sokolov A.N., Kuz'mina L.A., Klyasova G.A., Gaponova T.V., Baranova O.Yu., Lapin V.A., Konstantinova T.S., Samoilova O.S., Kaporskaya T.S., Shatokhin S.A. Klinicheskie rekomendatsii po diagnostike i lecheniyu ostrykh limfoblastnykh leikozov vzroslykh. Natsional'noe gematologicheskoe obshchestvo. 2014. 65 s.
- Wei A.H., Döhner H., Pocock C., Montesinos P., Afanasyev B., Dombret H., Ravandi F., Sayar H., Jang J.H., Porkka K., Selleslag D., Sandhu I., Turgut M., Giai V., Ofran Y., Kizil Çakar M., Botelho de Sousa A., Rybka J., Frairia C., Borin L., Beltrami G., Čermák J., Ossenkoppele G.J., La Torre I., Skikne B., Kumar K., Dong Q., Beach C.L., Roboz G.J., for the QUAZAR AML-001 Trial Investigators†. Oral Azacitidine Maintenance Therapy for Acute Myeloid Leukemia in First Remission. N Engl J Med. 2020; 383(26): 2526-37. https://doi.org/10.1056/NEJMoa2004444.
- Montesinos P., Recher C., Vives S., Zarzycka E., Wang J., Bertani G., Heuser M., Calado R.T., Schuh A.C., Yeh S.P., Daigle S.R., Hui J., Pandya S.S., Gianolio D.A., de Botton S., Döhner H. Ivosidenib and Azacitidine in IDH1-Mutated Acute Myeloid Leukemia. N Engl J Med. 2022; 386(16): 1519-31. https://doi.org/10.1056/NEJMoa2117344.
- Niemeyer C.M., Flotho C., Lipka D.B., Starý J., Rössig C., Baruchel A., Klingebiel T., Micalizzi C., Michel G., Nysom K., Rives S., Schmugge Liner M., Zecca M., Schönung M., Baumann I., Nöllke P., Benettaib B., Biserna N., Poon J., Simcock M., Patturajan M., Menezes D., Gaudy A., van den Heuvel-Eibrink M.M., Locatelli F. Response to upfront azacitidine in juvenile myelomonocytic leukemia in the AZA-JMML001 trial. Blood Adv. 2021; 5(14): 2901-8. https://doi.org/10.1182/bloodadvances.2020004144.
- Jabbour E., Issa J.P., Garcia-Manero G., Kantarjian H. Evolution of decitabine development: accomplishments, ongoing investigations, and future strategies. Cancer. 2008; 112(11): 2341-51. https://doi.org/10.1002/cncr.23463.
- Briski R., Garcia-Manero G., Kantarjian H., Ravandi F. The history of oral decitabine/cedazuridine and its potential role in acute myeloid leukemia. Ther Adv Hematol. 2023; 14. https://doi.org/10.1177/20406207231205429.
- Pollyea D.A., Winters A., McMahon C., Schwartz M., Jordan C.T., Rabinovitch R., Abbott D., Smith C.A., Gutman J.A. Venetoclax and azacitidine followed by allogeneic transplant results in excellent outcomes and may improve outcomes versus maintenance therapy among newly diagnosed AML patients older than 60. Bone Marrow Transplant. 2022; 57(2): 160-6. https://doi.org/10.1038/s41409-021-01476-7.
- Sekeres M.A., Watts J., Radinoff A., Sangerman M.A., Cerrano M., Lopez P.F., Zeidner J.F., Campelo M.D., Graux C., Liesveld J., Selleslag D., Tzvetkov N., Fram R.J., Zhao D., Bell J., Friedlander S., Faller D.V., Adès L. Randomized phase 2 trial of pevonedistat plus azacitidine versus azacitidine for higher-risk MDS/CMML or low-blast AML. Leukemia. 2021; 35(7): 2119-24. https://doi.org/10.1038/s41375-021-01125-4. Erratum in: Leukemia. 2021; 35(12): 3637. https://doi.org/10.1038/s41375-021-01473-1.
- DiNardo C.D., Schuh A.C., Stein E.M., Montesinos P., Wei A.H., de Botton S., Zeidan A.M., Fathi A.T., Kantarjian H.M., Bennett J.M., Frattini M.G., Martin-Regueira P., Lersch F., Gong J., Hasan M., Vyas P., Döhner H. Enasidenib plus azacitidine versus azacitidine alone in patients with newly diagnosed, mutant-IDH2 acute myeloid leukaemia (AG221- AML-005): a single-arm, phase 1b and randomised, phase 2 trial. Lancet Oncol. 2021; 22(11): 1597-608. https://doi.org/10.1016/S1470-2045(21)00494-0.
- Ohanian M., Garcia-Manero G., Levis M., Jabbour E., Daver N., Borthakur G., Kadia T., Pierce S., Burger J., Richie M.A., Patel K., Andreeff M., Estrov Z., Cortes J., Kantarjian H., Ravandi F. Sorafenib Combined with 5-azacytidine in Older Patients with Untreated FLT3-ITD Mutated Acute Myeloid Leukemia. Am J Hematol. 2018; 93(9): 1136-41. https://doi.org/10.1002/ajh.25198.
- Hu J., Wang X., Chen F., Ding M., Dong M., Yang W., Yin M., Wu J., Zhang L., Fu X., Sun Z., Li L., Wang X., Li X., Guo S., Zhang D., Lu X., Leng Q., Zhang M., Zhu L., Zhang X., Chen Q. Combination of Decitabine and a Modified Regimen of Cisplatin, Cytarabine and Dexamethasone: A Potential Salvage Regimen for Relapsed or Refractory Diffuse Large B-Cell Lymphoma After Second-Line Treatment Failure. Front Oncol. 2021; 11. https://doi.org/10.3389/fonc.2021.687374.
- Buocikova V., Tyciakova S., Pilalis E., Mastrokalou C., Urbanova M., Matuskova M., Demkova L., Medova V., Longhin E.M., Rundén-Pran E., Dusinska M., Rios-Mondragon I., Cimpan M.R., Gabelova A., Soltysova A., Smolkova B., Chatziioannou A. Decitabine-induced DNA methylationmediated transcriptomic reprogramming in human breast cancer cell lines; the impact of DCK overexpression. Front Pharmacol. 2022; 13. https://doi.org/10.3389/fphar.2022.991751.
- Champion C., Guianvarc’h D., Sénamaud-Beaufort C., Jurkowska R.Z., Jeltsch A., Ponger L., Arimondo P.B., Guieysse-Peugeot A.L. Mechanistic insights on the inhibition of c5 DNA methyltransferases by zebularine. PLoS One. 2010; 5(8). https://doi.org/10.1371/journal.pone.0012388.
- Lu Y., Chan Y.T., Tan H.Y., Li S., Wang N., Feng Y. Epigenetic regulation in human cancer: the potential role of epi-drug in cancer therapy. Mol Cancer. 2020; 19(1): 79. https://doi.org/10.1186/s12943-020-01197-3.
- Cheng J.C., Yoo C.B., Weisenberger D.J., Chuang J., Wozniak C., Liang G., Marquez V.E., Greer S., Orntoft T.F., Thykjaer T., Jones P.A. Preferential response of cancer cells to zebularine. Cancer Cell. 2004; 6(2): 151-8. https://doi.org/10.1016/j.ccr.2004.06.023.
- Takemura Y., Satoh M., Hatanaka K., Kubota S. Zebularine exerts its antiproliferative activity through S phase delay and cell death in human malignant mesothelioma cells. Biosci Biotechnol Biochem. 2018; 82(7): 1159-64. https://doi.org/10.1080/09168451.2018.1459466.
- Cheng J.C., Weisenberger D.J., Gonzales F.A., Liang G., Xu G.L., Hu Y.G., Marquez V.E., Jones P.A. Continuous zebularine treatment effectively sustains demethylation in human bladder cancer cells. Mol Cell Biol. 2004; 24(3): 1270-8. https://doi.org/10.1128/MCB.24.3.1270-1278.2004.
- Lemaire M., Momparler L.F., Raynal N.J., Bernstein M.L., Momparler R.L. Inhibition of cytidine deaminase by zebularine enhances the antineoplastic action of 5-aza-2'-deoxycytidine. Cancer Chemother Pharmacol. 2009; 63(3): 411-6. https://doi.org/10.1007/s00280-008-0750-6.
- Fulkerson C.M., Dhawan D., Jones D.R., Marquez V.E., Jones P.A., Wang Z., Wu Q., Klaunig J.E., Fourez L.M., Bonney P.L., Knapp D.W. Pharmacokinetics and toxicity of the novel oral demethylating agent zebularine in laboratory and tumor bearing dogs. Vet Comp Oncol. 2017; 15(1): 226-36. https://doi.org/10.1111/vco.12159.
- Holleran J.L., Eiseman J.L., Parise R.A., Kummar S., Beumer J.H. LC-MS/MS assay for the quantitation of FdCyd and its metabolites FdUrd and FU in human plasma. J Pharm Biomed Anal. 2016; 129: 359-66. https://doi.org/10.1016/j.jpba.2016.07.027.
- Guo D., Myrdal P.B., Karlage K.L., O’Connell S.P., Wissinger T.J., Tabibi S.E., Yalkowsky S.H. Stability of 5-fluoro-2'-deoxycytidine and tetrahydrouridine in combination. AAPS PharmSciTech. 2010; 11(1): 247-52. https://doi.org/10.1208/s12249-010-9383-2.
- Holleran J.L., Beumer J.H., McCormick D.L., Johnson W.D., Newman E.M., Doroshow J.H., Kummar S., Covey J.M., Davis M., Eiseman J.L. Oral and intravenous pharmacokinetics of 5-fluoro-2'-deoxycytidine and THU in cynomolgus monkeys and humans. Cancer Chemother Pharmacol. 2015; 76(4): 803-11. https://doi.org/10.1007/s00280-015-2857-x.
- Coyne G.O.', Wang L., Zlott J., Juwara L., Covey J.M., Beumer J.H., Cristea M.C., Newman E.M., Koehler S., Nieva J.J., Garcia A.A., Gandara D.R.,Miller B., Khin S., Miller S.B., Steinberg S.M., Rubinstein L., Parchment R.E., Kinders R.J., Piekarz R.L., Kummar S., Chen A.P., Doroshow J.H. Intravenous 5-fluoro-2’-deoxycytidine administered with tetrahydrouridine increases the proportion of p16-expressing circulating tumor cells in patients with advanced solid tumors. Cancer Chemother Pharmacol. 2020; 85(5): 979-93. https://doi.org/10.1007/s00280-020-04073-5.
- Brueckner B., Rius M., Markelova M.R., Fichtner I., Hals P.A., Sandvold M.L., Lyko F. Delivery of 5-azacytidine to human cancer cells by elaidic acid esterification increases therapeutic drug efficacy. Mol Cancer Ther. 2010; 9(5): 1256-64. https://doi.org/10.1158/1535-7163.MCT-09-1202.
- Rius M., Stresemann C., Keller D., Brom M., Schirrmacher E., Keppler D., Lyko F. Human concentrative nucleoside transporter 1-mediated uptake of 5-azacytidine enhances DNA demethylation. Mol Cancer Ther. 2009; 8(1): 225-31. https://doi.org/10.1158/1535-7163.MCT-08-0743.
- Byun H.M., Choi S.H., Laird P.W., Trinh B., Siddiqui M.A., Marquez V.E., Yang A.S. 2'-Deoxy-N4-[2-(4-nitrophenyl)ethoxycarbonyl]-5 -azacytidine: a novel inhibitor of DNA methyltransferase that requires activation by human carboxylesterase 1. Cancer Lett. 2008; 266(2): 238-48. https://doi.org/10.1016/j.canlet.2008.02.069.
- Srivastava P., Paluch B.E., Matsuzaki J., James S.R., CollamatLai G., Karbach J., Nemeth M.J., Taverna P., Karpf A.R., Griffiths E.A. Immunomodulatory action of SGI-110, a hypomethylating agent, in acute myeloid leukemia cells and xenografts. Leuk Res. 2014; 38(11): 1332-41. https://doi.org/10.1016/j.leukres.2014.09.001.
- Garcia-Manero G., Roboz G., Walsh K., Kantarjian H., Ritchie E., Kropf P., O’Connell C., Tibes R., Lunin S., Rosenblat T., Yee K., Stock W., Griffiths E., Mace J., Podoltsev N., Berdeja J., Jabbour E., Issa J.J., Hao Y., Keer H.N., Azab M., Savona M.R. Guadecitabine (SGI-110) in patients with intermediate or high-risk myelodysplastic syndromes: phase 2 results from a multicentre, open-label, randomised, phase 1/2 trial. Lancet Haematol. 2019; 6(6): 317-27. https://doi.org/10.1016/S2352-3026(19)30029-8.
- Oza A.M., Matulonis U.A., Secord A.A., Nemunaitis J., Roman L.D., Blagden S.P., Banerjee S., McGuire W.P., Ghamande S., Birrer M.J., Fleming G.F., Markham M.J., Hirte H.W., Provencher D.M., Basu B., Kristeleit R., Armstrong D.K., Schwartz B., Braly P., Hall G.D., Nephew K.P., Jueliger S., Oganesian A., Naim S., Hao Y., Keer H., Azab M., Matei D. A Randomized Phase II Trial of Epigenetic Priming with Guadecitabine and Carboplatin in Platinum-resistant, Recurrent Ovarian Cancer. Clin Cancer Res. 2020; 26(5): 1009-16. https://doi.org/10.1158/1078-0432.CCR-19-1638.
- Chen S., Xie P., Cowan M., Huang H., Cardenas H., Keathley R., Tanner E.J., Fleming G.F., Moroney J.W., Pant A., Akasha A.M., Davuluri R.V., Kocherginsky M., Zhang B., Matei D. Epigenetic priming enhances antitumor immunity in platinum-resistant ovarian cancer. J Clin Invest. 2022; 132(14). https://doi.org/10.1172/JCI158800.
- Crabb S.J., Danson S., Catto J.W.F., Hussain S., Chan D., Dunkley D., Downs N., Marwood E., Day L., Saunders G., Light M., Whitehead A., Ellis D., Sarwar N., Enting D., Birtle A., Johnson B., Huddart R., Griffiths G. Phase I Trial of DNA Methyltransferase Inhibitor Guadecitabine Combined with Cisplatin and Gemcitabine for Solid Malignancies Including Urothelial Carcinoma (SPIRE). Clin Cancer Res. 2021; 27(7): 1882-92. https://doi.org/10.1158/1078-0432.CCR-20-3946.
- Brueckner B., Lyko F. DNA methyltransferase inhibitors: old and new drugs for an epigenetic cancer therapy. Trends Pharmacol Sci. 2004; 25(11): 551-4. https://doi.org/10.1016/j.tips.2004.09.004.
- Ou Y., Zhang Q., Tang Y., Lu Z., Lu X., Zhou X., Liu C. DNA methylation enzyme inhibitor RG108 suppresses the radioresistance of esophageal cancer. Oncol Rep. 2018; 39(3): 993-1002. https://doi.org/10.3892/or.2018.6210.
- Yang L., Hou J., Cui X.H., Suo L.N., Lv Y.W. RG108 induces the apoptosis of endometrial cancer Ishikawa cell lines by inhibiting the expression of DNMT3B and demethylation of HMLH1. Eur Rev Med Pharmacol Sci. 2017; 21(22): 5056-64. https://doi.org/10.26355/eurrev_201711_13818.
- Lee B.H., Yegnasubramanian S., Lin X., Nelson W.G. Procainamide is a specific inhibitor of DNA methyltransferase 1. J Biol Chem. 2005; 280(49): 40749-56. https://doi.org/10.1074/jbc.M505593200.
- Villar-Garea A., Fraga M.F., Espada J., Esteller M. Procaine is a DNA-demethylating agent with growth-inhibitory effects in human cancer cells. Cancer Res. 2003; 63(16): 4984-9.
- Sabit H., Samy M.B., Said O.A., El-Zawahri M.M. Procaine Induces Epigenetic Changes in HCT116 Colon Cancer Cells. Genet Res Int. 2016. https://doi.org/10.1155/2016/8348450.
- Li Y.C., Wang Y., Li D.D., Zhang Y., Zhao T.C., Li C.F. Procaine is a specific DNA methylation inhibitor with anti-tumor effect for human gastric cancer. J Cell Biochem. 2018; 119(2): 2440-9. https://doi.org/10.1002/jcb.26407.
- Ma X.W., Li Y., Han X.C., Xin Q.Z. The effect of low dosage of procaine on lung cancer cell proliferation. Eur Rev Med Pharmacol Sci. 2016; 20(22): 4791-5.
- Gao Z., Xu Z., Hung M.S., Lin Y.C., Wang T., Gong M., Zhi X., Jablons D.M., You L. Procaine and procainamide inhibit the Wnt canonical pathway by promoter demethylation of WIF-1 in lung cancer cells. Oncol Rep. 2009; 22(6): 1479-84. https://doi.org/10.3892/or_00000590.
- Uetrecht J.P., Freeman R.W., Woosley R.L. The implications of procainamide metabolism to its induction of lupus. Arthritis Rheum. 1981; 24(8): 994-1003. https://doi.org/10.1002/art.1780240803.
- Paşa S., Erdogan O., Cevik O. Design, synthesis and investigation of procaine based new Pd complexes as DNA methyltransferase inhibitor on gastric cancer cells. Inorg Chem Comm. 2021; 132. https://doi.org/10.1016/j.inoche.2021.108846.
- Tanaka H., Marumo H., Nagai T., Okada M., Taniguchi K. Nanaomycins, new antibiotics produced by a strain of Streptomyces. III. A new component, nanaomycin C, and biological activities of nanaomycin derivatives. J Antibiot (Tokyo). 1975; 28(12): 925-30. https://doi.org/10.7164/antibiotics.28.925.
- Kormanec J., Novakova R., Csolleiova D., Feckova L., Rezuchova B., Sevcikova B., Homerova D. The antitumor antibiotic mithramycin: new advanced approaches in modification and production. Appl Microbiol Biotechnol. 2020; 104(18): 7701-21. https://doi.org/10.1007/s00253-020-10782-x.
- Kuck D., Caulfield T., Lyko F., Medina-Franco J.L. Nanaomycin A selectively inhibits DNMT3B and reactivates silenced tumor suppressor genes in human cancer cells. Mol Cancer Ther. 2010; 9(11): 3015-23. https://doi.org/10.1158/1535-7163.MCT-10-0609.
- Liu P.Y., Sokolowski N., Guo S.T., Siddiqi F., Atmadibrata B., Telfer T.J., Sun Y., Zhang L., Yu D., Mccarroll J., Liu B., Yang R.H., Guo X.Y., Tee A.E., Itoh K., Wang J., Kavallaris M., Haber M., Norris M.D., Cheung B.B., Byrne J.A., Ziegler D.S., Marshall G.M., Dinger M.E., Codd R., Zhang X.D., Liu T. The BET bromodomain inhibitor exerts the most potent synergistic anticancer effects with quinone-containing compounds and anti-microtubule drugs. Oncotarget. 2016; 7(48): 79217-32. https://doi.org/10.18632/oncotarget.12640.
- Lin R.K., Hsu C.H., Wang Y.C. Mithramycin A inhibits DNA methyltransferase and metastasis potential of lung cancer cells. Anticancer Drugs. 2007; 18(10): 1157-64. https://doi.org/10.1097/CAD.0b013e3282a215e9.
- Arce C., Segura-Pacheco B., Perez-Cardenas E., Taja-Chayeb L., Candelaria M., Dueñnas-Gonzalez A. Hydralazine target: from blood vessels to the epigenome. J Transl Med. 2006; 4: 10. https://doi.org/10.1186/1479-5876-4-10.
- Graça I., Sousa E.J., Costa-Pinheiro P., Vieira F.Q., TorresFerreira J., Martins M.G., Henrique R., Jerónimo C. Anti-neoplastic properties of hydralazine in prostate cancer. Oncotarget. 2014; 5(15): 5950-64. https://doi.org/10.18632/oncotarget.1909.
- Singh N., Dueñas-González A., Lyko F., Medina-Franco J.L. Molecular modeling and molecular dynamics studies of hydralazine with human DNA methyltransferase 1. ChemMedChem. 2009; 4(5): 792-9. https://doi.org/10.1002/cmdc.200900017.
- Kumanishi S., Yamanegi K., Nishiura H., Fujihara Y., Kobayashi K., Nakasho K., Futani H., Yoshiya S. Epigenetic modulators hydralazine and sodium valproate act synergistically in VEGI-mediated anti-angiogenesis and VEGF interference in human osteosarcoma and vascular endothelial cells. Int J Oncol. 2019; 55(1): 167-78. https://doi.org/10.3892/ijo.2019.4811.
- Bauman J., Shaheen M., Verschraegen C.F., Belinsky S.A., Houman Fekrazad M., Lee F.C., Rabinowitz I., Ravindranathan M., Jones D.V. Jr. A Phase I Protocol of Hydralazine and Valproic Acid in Advanced, Previously Treated Solid Cancers. Transl Oncol. 2014; 7(3): 349-54. https://doi.org/10.1016/j.tranon.2014.03.001.
- Espinoza-Zamora J.R., Labardini-Méndez J., Sosa-Espinoza A., López-González C., Vieyra-García M., Candelaria M., Lozano-Zavaleta V., Toledano-Cuevas D.V., Zapata-Canto N., Cervera E., Dueñas-González A. Efficacy of hydralazine and valproate in cutaneous T-cell lymphoma, a phase II study. Expert Opin Investig Drugs. 2017; 26(4): 481-7. https://doi.org/10.1080/13543784.2017.1291630. Erratum in: Expert Opin Investig Drugs. 2017; 26(4): 523. https://doi.org/10.1080/13543784.2017.1306178.
- Maiti A., Daver N.G. Eprenetapopt in the Post-Transplant Setting: Mechanisms and Future Directions. J Clin Oncol. 2022; 40(34): 3994-7. https://doi.org/10.1200/JCO.22.01505.
- Qiang W., Jin T., Yang Q., Liu W., Liu S., Ji M., He N., Chen C., Shi B., Hou P. PRIMA-1 selectively induces global DNA demethylation in p53 mutant-type thyroid cancer cells. J Biomed Nanotechnol. 2014; 10(7): 1249-58. https://doi.org/10.1166/jbn.2014.1862.
- Teoh P.J., Bi C., Sintosebastian C., Tay L.S., Fonseca R., Chng W.J. PRIMA-1 targets the vulnerability of multiple myeloma of deregulated protein homeostasis through the perturbation of ER stress via p73 demethylation. Oncotarget. 2016; 7(38): 61806-19. https://doi.org/10.18632/oncotarget.11241.
- Fujihara K.M., Zhang B.Z., Jackson T.D., Ogunkola M.O., Nijagal B., Milne J.V., Sallman D.A., Ang C.S., Nikolic I., Kearney C.J., Hogg S.J., Cabalag C.S., Sutton V.R., Watt S., Fujihara A.T., Trapani J.A., Simpson K.J., Stojanovski D., Leimkühler S., Haupt S., Phillips W.A., Clemons N.J. Eprenetapopt triggers ferroptosis, inhibits NFS1 cysteine desulfurase, and synergizes with serine and glycine dietary restriction. Sci Adv. 2022; 8(37). https://doi.org/10.1126/sciadv.abm9427.
- Amirtharaj F., Venkatesh G.H., Wojtas B., Nawafleh H.H., Mahmood A.S., Nizami Z.N., Khan M.S., Thiery J., Chouaib S. p53 reactivating small molecule PRIMA-1MET/APR-246 regulates genomic instability in MDA-MB-231 cells. Oncol Rep. 2022; 47(4): 85. https://doi.org/10.3892/or.2022.8296.
- Fransson Å., Glaessgen D., Alfredsson J., Wiman K.G., BajalicaLagercrantz S., Mohell N. Strong synergy with APR-246 and DNAdamaging drugs in primary cancer cells from patients with TP53 mutant High-Grade Serous ovarian cancer. J Ovarian Res. 2016; 9(1): 27. https://doi.org/10.1186/s13048-016-0239-6.
- Sallman D.A., Dezern A.E., Steensma D., Sweet K.L., Cluzeau T., Sekeres M., Garcia-Manero G., Roboz G.J., McLemore A.F., McGraw K.L., Puskas J., Zhang L., Bhagat C.K., Yao J., Ali N.A., Padron E., Tell R., Lancet J.E., Fenaux P., List A., Komrokji R.S. Phase 1b/2 Combination Study of APR-246 and Azacitidine (AZA) in Patients with TP53 mutant Myelodysplastic Syndromes (MDS) and Acute Myeloid Leukemia (AML). Blood Adv. 2018; 132 (s1). https://doi.org/10.1182/blood-2018-99-119990.
- Sun N., Zhang J., Zhang C., Zhao B., Jiao A. DNMTs inhibitor SGI-1027 induces apoptosis in Huh7 human hepatocellular carcinoma cells. Oncol Lett. 2018; 16(5): 5799-806. https://doi.org/10.3892/ol.2018.9390.
- García-Domínguez P., Dell'aversana C., Alvarez R., Altucci L., de Lera A.R. Synthetic approaches to DNMT inhibitor SGI-1027 and effects on the U937 leukemia cell line. Bioorg Med Chem Lett. 2013; 23(6): 1631-5. https://doi.org/10.1016/j.bmcl.2013.01.085.
- Kelly G.S. Quercetin. Monograph. Altern Med Rev. 2011; 16(2): 172-94.
- Bilyk O.V., Rybal'chenko V.K., Romanyuk B.P. Bioflavonoid kvertsetin i perspektivy ego ispol'zovaniya v meditsine. Zagal'na patologiya ta patologichna fiziologiya. 2007; 2(1): 4-9.
- Alvarez M.C., Maso V., Torello C.O., Ferro K.P., Saad S.T.O. The polyphenol quercetin induces cell death in leukemia by targeting epigenetic regulators of pro-apoptotic genes. Clin Epigenetics. 2018; 10(1): 139. https://doi.org/10.1186/s13148-018-0563-3.
- Kedhari Sundaram M., Hussain A., Haque S., Raina R., Afroze N. Quercetin modifies 5’CpG promoter methylation and reactivates various tumor suppressor genes by modulating epigenetic marks in human cervical cancer cells. J Cell Biochem. 2019; 120(10): 18357-69. https://doi.org/10.1002/jcb.29147.
- Almatroodi S.A., Almatroudi A., Khan A.A., Alhumaydhi F.A., Alsahli M.A., Rahmani A.H. Potential Therapeutic Targets of Epigallocatechin Gallate (EGCG), the Most Abundant Catechin in Green Tea, and Its Role in the Therapy of Various Types of Cancer. Molecules. 2020; 25(14): 3146. https://doi.org/10.3390/molecules25143146.
- Minnelli C., Cianfruglia L., Laudadio E., Mobbili G., Galeazzi R., Armeni T. Effect of Epigallocatechin-3-Gallate on EGFR Signaling and Migration in Non-Small Cell Lung Cancer. Int J Mol Sci. 2021; 22(21). https://doi.org/10.3390/ijms222111833.
- Della Via F.I., Shiraishi R.N., Santos I., Ferro K.P., SalazarTerreros M.J., Franchi Junior G.C., Rego E.M., Saad S.T.O., Torello C.O. (-)-Epigallocatechin-3-gallate induces apoptosis and differentiation in leukaemia by targeting reactive oxygen species and PIN1. Sci Rep. 2021; 11(1). https://doi.org/10.1038/s41598-021-88478-z.
- Sheng J., Shi W., Guo H., Long W., Wang Y., Qi J., Liu J., Xu Y. The Inhibitory Effect of (-)-Epigallocatechin-3-Gallate on Breast Cancer Progression via Reducing SCUBE2 Methylation and DNMT Activity. Molecules. 2019; 24(16). https://doi.org/10.3390/molecules24162899.
- Khan M.A., Hussain A., Sundaram M.K., Alalami U., Gunasekera D., Ramesh L., Hamza A., Quraishi U. (-)-Epigallocatechin-3-gallate reverses the expression of various tumor-suppressor genes by inhibiting DNA methyltransferases and histone deacetylases in human cervical cancer cells. Oncol Rep. 2015; 33(4): 1976-84. https://doi.org/10.3892/or.2015.3802.
- Nandakumar V., Vaid M., Katiyar S.K. (-)-Epigallocatechin3-gallate reactivates silenced tumor suppressor genes, Cip1/p21 and p16INK4a, by reducing DNA methylation and increasing histones acetylation in human skin cancer cells. Carcinogenesis. 2011; 32(4): 537-44. https://doi.org/10.1093/carcin/bgq285.
- Alizadeh M., Nafari A., Safarzadeh A., Veiskarami S., Almasian M., Asghar Kiani A. The Impact of EGCG and RG108 on SOCS1 Promoter DNA Methylation and Expression in U937 Leukemia Cells. Rep Biochem Mol Biol. 2021; 10(3): 455-61. https://doi.org/10.52547/rbmb.10.3.455.
- Khan M.I., Nur S.M., Abdulaal W.H. A study on DNA methylation modifying natural compounds identified EGCG for induction of IFI16 gene expression related to the innate immune response in cancer cells. Oncol Lett. 2022; 24(1): 218. https://doi.org/10.3892/ol.2022.13339.
- Fang M.Z., Wang Y., Ai N., Hou Z., Sun Y., Lu H., Welsh W., Yang C.S. Tea polyphenol (-)-epigallocatechin-3-gallate inhibits DNA methyltransferase and reactivates methylation-silenced genes in cancer cell lines. Cancer Res. 2003; 63(22): 7563-70.
- Chen L.L., Han W.F., Geng Y., Su J.S. A genome-wide study of DNA methylation modified by epigallocatechin-3-gallate in the CAL-27 cell line. Mol Med Rep. 2015; 12(4): 5886-90. https://doi.org/10.3892/mmr.2015.4118.
- McLarty J., Bigelow R.L., Smith M., Elmajian D., Ankem M., Cardelli J.A. Tea polyphenols decrease serum levels of prostate-specific antigen, hepatocyte growth factor, and vascular endothelial growth factor in prostate cancer patients and inhibit production of hepatocyte growth factor and vascular endothelial growth factor in vitro. Cancer Prev Res (Phila). 2009; 2(7): 673-82. https://doi.org/10.1158/1940-6207.CAPR-08-0167.
- Jazvinšćak Jembrek M., Oršolić N., Mandić L., Sadžak A., Šegota S. Anti-Oxidative, Anti-Inflammatory and Anti-Apoptotic Effects of Flavonols: Targeting Nrf2, NF-κB and p53 Pathways in Neurodegeneration. Antioxidants (Basel). 2021; 10(10). https://doi.org/10.3390/antiox10101628.
- Jia S., Xu X., Zhou S., Chen Y., Ding G., Cao L. Fisetin induces autophagy in pancreatic cancer cells via endoplasmic reticulum stress- and mitochondrial stress-dependent pathways. Cell Death Dis. 2019; 10(2). https://doi.org/10.1038/s41419-019-1366-y. Erratum in: Cell Death Dis. 2024; 15(1). https://doi.org/10.1038/s41419-023-06399-3.
- Mukhtar E., Adhami V.M., Sechi M., Mukhtar H. Dietary flavonoid fisetin binds to β-tubulin and disrupts microtubule dynamics in prostate cancer cells. Cancer Lett. 2015; 367(2): 173-83. https://doi.org/10.1016/j.canlet.2015.07.030.
- Hassan F.U., Rehman M.S., Khan M.S., Ali M.A., Javed A., Nawaz A., Yang C. Curcumin as an Alternative Epigenetic Modulator: Mechanism of Action and Potential Effects. Front Genet. 2019; 10. https://doi.org/10.3389/fgene.2019.00514.
- Kirsanov K.I., Vlasova O.A., Fetisov T.I., Zenkov R.G., Lesovaya E.A., Belitskii G.A., Gurova K., Yakubovskaya M.G. Vliyanie DNKtropnykh antikantserogennykh soedinenii na mekhanizmy regulyatsii ekspressii genov. Uspekhi molekulyarnoi onkologii. 2018; 5(4): 41-63. https://doi.org/10.17650/2313-805X-2018-5-4-41-63.
- Yu J., Peng Y., Wu L.C., Xie Z., Deng Y., Hughes T., He S., Mo X., Chiu M., Wang Q.E., He X., Liu S., Grever M.R., Chan K.K., Liu Z. Curcumin down-regulates DNA methyltransferase 1 and plays an anti-leukemic role in acute myeloid leukemia. PLoS One. 2013; 8(2). https://doi.org/10.1371/ journal.pone.0055934.
- Chen J., Ying Y., Zhu H., Zhu T., Qu C., Jiang J., Fang B. Curcumininduced promoter hypermethylation of the mammalian target of rapamycin gene in multiple myeloma cells. Oncol Lett. 2019; 17(1): 1108-14. https://doi.org/10.3892/ol.2018.9662.
- Al-Yousef N., Shinwari Z., Al-Shahrani B., Al-Showimi M., AlMoghrabi N. Curcumin induces re-expression of BRCA1 and suppression of γ synuclein by modulating DNA promoter methylation in breast cancer cell lines. Oncol Rep. 2020; 43(3): 827-38. https://doi.org/10.3892/or.2020.7473.
- Link A., Balaguer F., Shen Y., Lozano J.J., Leung H.C., Boland C.R., Goel A. Curcumin modulates DNA methylation in colorectal cancer cells. PLoS One. 2013; 8(2). https://doi.org/10.1371/journal.pone.0057709.
- Hosokawa M., Seiki R., Iwakawa S., Ogawara K.I. Combination of azacytidine and curcumin is a potential alternative in decitabineresistant colorectal cancer cells with attenuated deoxycytidine kinase. Biochem Biophys Res Commun. 2021; 578: 157-62. https://doi.org/10.1016/j.bbrc.2021.09.041.
- Howells L.M., Iwuji C.O.O., Irving G.R.B., Barber S., Walter H., Sidat Z., Griffin-Teall N., Singh R., Foreman N., Patel S.R., Morgan B., Steward W.P., Gescher A., Thomas A.L., Brown K. Curcumin Combined with FOLFOX Chemotherapy Is Safe and Tolerable in Patients with Metastatic Colorectal Cancer in a Randomized Phase IIa Trial. J Nutr. 2019; 149(7): 1133-9. https://doi.org/10.1093/jn/nxz029.
- Saghatelyan T., Tananyan A., Janoyan N., Tadevosyan A., Petrosyan H., Hovhannisyan A., Hayrapetyan L., Arustamyan M., Arnhold J., Rotmann A.R., Hovhannisyan A., Panossian A. Efficacy and safety of curcumin in combination with paclitaxel in patients with advanced, metastatic breast cancer: A comparative, randomized, double-blind, placebo-controlled clinical trial. Phytomedicine. 2020; 70. https://doi.org/10.1016/j.phymed.2020.153218.
- Rauf A., Imran M., Suleria H.A.R., Ahmad B., Peters D.G., Mubarak M.S. A comprehensive review of the health perspectives of resveratrol. Food Funct. 2017; 8(12): 4284-305. https://doi.org/10.1039/c7fo01300k.
- Aldawsari F.S., Aguayo-Ortiz R., Kapilashrami K., Yoo J., Luo M., Medina-Franco J.L., Velázquez-Martínez C.A. Resveratrolsalicylate derivatives as selective DNMT3 inhibitors and anticancer agents. J Enzyme Inhib Med Chem. 2016; 31(5): 695-703. https://doi.org/10.3109/14756366.2015.1058256.
- Izquierdo-Torres E., Hernández-Oliveras A., Meneses-Morales I., Rodríguez G., Fuentes-García G., Zarain-Herzberg Á. Resveratrol upregulates ATP2A3 gene expression in breast cancer cell lines through epigenetic mechanisms. Int J Biochem Cell Biol. 2019; 113: 37-47. https://doi.org/10.1016/j.biocel.2019.05.020.
- Sharifi-Rad J., Quispe C., Imran M., Rauf A., Nadeem M., Gondal T.A., Ahmad B., Atif M., Mubarak M.S., Sytar O., Zhilina O.M., Garsiya E.R., Smeriglio A., Trombetta D., Pons D.G., Martorell M., Cardoso S.M., Razis A.F.A., Sunusi U., Kamal R.M., Rotariu L.S., Butnariu M., Docea A.O., Calina D. Genistein: An Integrative Overview of Its Mode of Action, Pharmacological Properties, and Health Benefits. Oxid Med Cell Longev. 2021. https://doi.org/10.1155/2021/3268136.
- Sundaram M.K., Ansari M.Z., Al Mutery A., Ashraf M., Nasab R., Rai S., Rais N., Hussain A. Genistein Induces Alterations of Epigenetic Modulatory Signatures in Human Cervical Cancer Cells. Anticancer Agents Med Chem. 2018; 18(3): 412-21. https://doi.org/10.2174/1871520617666170918142114.
- Sharma M., Tollefsbol T.O. Combinatorial epigenetic mechanisms of sulforaphane, genistein and sodium butyrate in breast cancer inhibition. Exp Cell Res. 2022; 416(1). https://doi.org/10.1016/j.yexcr.2022.113160.
- Xie Q., Bai Q., Zou L.Y., Zhang Q.Y., Zhou Y., Chang H., Yi L., Zhu J.D., Mi M.T. Genistein inhibits DNA methylation and increases expression of tumor suppressor genes in human breast cancer cells. Genes Chromosomes Cancer. 2014; 53(5): 422-31. https://doi.org/10.1002/gcc.22154.
- Romagnolo D.F., Donovan M.G., Papoutsis A.J., Doetschman T.C., Selmin O.I. Genistein Prevents BRCA1 CpG Methylation and Proliferation in Human Breast Cancer Cells with Activated Aromatic Hydrocarbon Receptor. Curr Dev Nutr. 2017; 1(6). https://doi.org/10.3945/cdn.117.000562.
- Li H., Xu W., Huang Y., Huang X., Xu L., Lv Z. Genistein demethylates the promoter of CHD5 and inhibits neuroblastoma growth in vivo. Int J Mol Med. 2012; 30(5): 1081-6. https://doi.org/10.3892/ijmm.2012.1118.
- Pintova S., Dharmupari S., Moshier E., Zubizarreta N., Ang C., Holcombe R.F. Genistein combined with FOLFOX or FOLFOX-Bevacizumab for the treatment of metastatic colorectal cancer: phase I/II pilot study. Cancer Chemother Pharmacol. 2019; 84(3): 591-8. https://doi.org/10.1007/s00280-019-03886-3.
- Chua G.N.L., Wassarman K.L., Sun H., Alp J.A., Jarczyk E.I., Kuzio N.J., Bennett M.J., Malachowsky B.G., Kruse M., Kennedy A.J. Cytosine-Based TET Enzyme Inhibitors. ACS Med Chem Lett. 2019; 10(2): 180-5. https://doi.org/10.1021/acsmedchemlett.8b00474.
- Weirath N.A., Hurben A.K., Chao C., Pujari S.S., Cheng T., Liu S., Tretyakova N.Y. Small Molecule Inhibitors of TET Dioxygenases: Bobcat339 Activity Is Mediated by Contaminating Copper(II). ACS Med Chem Lett. 2022; 13(5): 792-8. https://doi.org/10.1021/acsmedchemlett.1c00677.
- Singh A.K., Zhao B., Liu X., Wang X., Li H., Qin H., Wu X., Ma Yu., Horne D., Yu X. Selective targeting of TET catalytic domain promotes somatic cell reprogramming. Proc Natl Acad Sci U S A. 2020; 117(7): 3621-6. https://doi.org/10.1073/pnas.1910702117.
- Guan Y., Tiwari A.D., Phillips J.G., Hasipek M., Grabowski D.R., Pagliuca S., Gopal P., Kerr C.M., Adema V., Radivoyevitch T., Parker Y., Lindner D.J., Meggendorfer M., Abazeed M., Sekeres M.A., Mian O.Y., Haferlach T., Maciejewski J.P., Jha B.K. A Therapeutic Strategy for Preferential Targeting of TET2 Mutant and TET-dioxygenase Deficient Cells in Myeloid Neoplasms. Blood Cancer Discov. 2021; 2(2): 146-61. https://doi.org/10.1158/2643-3230.BCD-20-0173.
