Получение порошков SnO2 разложением термически нестабильных соединений

Автор: Иванов В.В., Сидорак И.А., Шубин А.А., Денисова Л.Т.
Журнал: Журнал Сибирского федерального университета. Серия: Техника и технологии @technologies-sfu
Статья в выпуске: 2 т.3, 2010 года.
Бесплатный доступ
Дан обзор работ, посвященных использованию и изучению процессов получения SnO2 разложением термически нестабильных соединений - гидроксидов и солей. Представлены также результаты исследований авторов статьи в области указанных процессов при термическом разложении сульфата олова (II) методами термогравиметрии и дифференциальной сканирующей калориметрии.
Диоксид олова, порошки, термическое разложение, гидроксиды олова, соли олова, контактный материал
Короткий адрес: https://sciup.org/146114532
IDR: 146114532 | УДК: 621.762.214:546.814-31
Synthesis of SnO2 powders by decomposition of the thermally unstable compounds
The survey of the works dealt with the usage and study of the synthesis processes of SnO2 by the thermal decomposition of the unstable compounds - hydroxides and salts was held. Also, the authors results of the study of the listed processes in thermal decomposition of tin sulphate (II) by the methods thermogravimetrical analysis and differential scanning calorimetry are represented.
Текст научной статьи Получение порошков SnO2 разложением термически нестабильных соединений
Диоксид олова – соединение, имеющее весьма обширную область практического использования: прозрачные и электропроводящие пленки различного назначения, газовые сенсоры, катализаторы, электроды, функциональные композиционные материалы и др. [1-6]. В соответствии с назначением применяют разнообразные технологии изготовления таких материалов в виде нанодисперсных форм, порошков, компактной керамики, свойства которых в значительной степени зависят от способа получения оксида [7-12]. В ряде случаев традиционного применения компактной керамики целесообразно пользоваться промышленными микропорошками SnO2. Современные же области использования SnO2 связаны с высокодисперсными продуктами в виде нанопорошков, а также тонких пленок, и здесь наиболее распространенными методами получения являются жидкофазные методы мягкой химии: химическое осаждение, золь-гель-метод с различными вариациями и т.п. [13-20], имеющие в своей схеме, как правило, стадии разложения термически нестабильных соединений олова.
Олово в большинстве случаев принимает две возможные степени окисления: (II) и (IV). С учетом очевидных требований к свойствам прекурсора для синтеза оксида путем термического разложения (низкие скорости сублимации и испарения, адекватная целевому процессу температура разложения и отсутствие нежелательных продуктов, доступность и приемлемая
стоимость) удобные для использования соединения олова весьма немногочисленны. Особенно это справедливо в отношении соединений олова (IV). Наибольшее применение в рассматриваемом направлении нашли, по-видимому, гидроксиды олова и соли некоторых органических кислот, а также сульфаты. Гидроксиды являются, как правило, промежуточными продуктами в указанных выше методах мягкой химии, которые далее подвергаются термолизу с целью превращения в SnO2. Распылительный пиролиз - также один из самых распространенных способов синтеза SnO2, вследствие своей простоты и технологичности [21-28]. Прокаливание гидроксидов и оксалата – традиционный способ получения диоксида олова [29].
Таким образом, в основе многих методов синтеза этого оксида лежат процессы разложения термически нестабильных соединений. В то же время литературные данные по разложению соединений олова ограниченны и часто противоречивы. Особенно это касается применения соединений олова (II), где проблемы могут быть связаны с высоким восстановительным потенциалом в растворах и возможностью образования монооксида SnO при термолизе соответствующих соединений, особенно в инертной среде. Эти вопросы требуют специального рассмотрения и анализа, как с общих позиций, так и в каждом конкретном применении. Большое число работ посвящено изучению процессов получения тонких пленок SnO2. Мы ограничимся, в основном, рассмотрением получения порошковых форм, которые могут быть применены в процессах синтеза композиционных материалов, например, для разрывных электрических контактов.
Получение порошковых форм SnO2 из гидроксидов
Допированный железом наноразмерный порошок диоксида олова получали методом химического соосаждения [17] в приложении к задаче синтеза электроконтактного материала Ag/ SnO 2 . Это простой, дешевый и широко используемый в промышленности метод для сложных оксидных порошков. Допирование рассматривали как способ предотвращения роста кристаллитов во время синтеза. Химическое осаждение сложного гидроксида SnFe(OH)x проводили из водного раствора тетрахлорида олова SnCl 4 - 5H2O и хлорного железа (в мольном соотношении 14:5 и 4:5) гидратом аммиака при рН=8. Осадок промывали, сушили при 373 К и растирали в порошок. Чистые гидроксиды Sn(OH) 4 и Fe(OH)3 также готовили для сравнения. Продукты осаждения тестировали методами термогравиметрии (ТГ), дифференциальной сканирующей калориметрии (ДСК), рентгенофазовым анализом и просвечивающей электронной микроскопией (ПЭМ).
На рис. 1 показаны соответствующие ТГ- и ДСК-кривые для Sn(OH) 4 . Первый эндотермический пик на ДСК-кривой авторы связывают с удалением несвязанной адсорбированной воды, второй 618,5 К [345,7 + 273,15 = 618,9 К] (345,7 ° С) - с разложением гидроксида. Литературных данных для сравнения в работе не приведено. Известно, однако [30], что связанная вода трудно удаляется из SnO2, являющегося продуктом термолиза оловянных кислот. Результаты ТГ-анализа показывают, что стабилизация массы образца на уровне 80,82 % от исходной наблюдается лишь к температуре около 1270 К. Приведенное значение убыли массы очень близко к величине 80,71 %, рассчитанной по уравнению
Sn(OH) 4 ^ SnO 2 + 2Н 2 О.
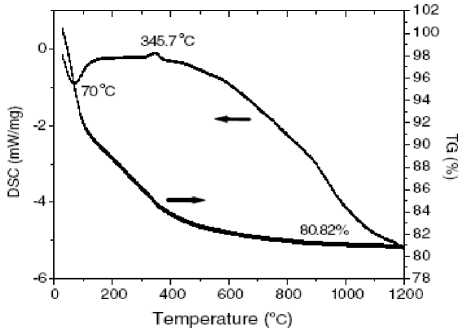
Рис. 1. ТГ- и ДСК-кривые для Sn(OH) 4 [17]
Поэтому объяснение результатов термического анализа, приводимое авторами, следует признать недостаточным. Реальный процесс сложнее и, по-видимому, может быть объяснен первичным разложением гидроксида при температуре около 343 К с образованием α-оловянной кислоты SnO2 ⋅ xH2O (1<х ≤ 2). Последующее нагревание приводит к постепенному удалению воды, образованию β-оловянной кислоты (х<1) и дальнейшему ее постепенному обезвоживанию. Обе оловянные кислоты имеют переменное содержание воды SnO2 ⋅ xH2O и теряют воду непрерывно [30].
Отжиг Sn(OH)4 в течение часа (рис. 2а) показывает, что кристаллизация начинается ниже 670 К, а типичные кристаллиты SnO 2 с тетрагональной структурой могут быть получены при температуре 823 К. Сложный гидроксид (рис. 2б) кристаллизуется при более высокой температуре 923 К. Выше 1023 К начинает выделяться фаза Fe 2 O 3 .
При температуре выше 823 К для Sn(OH)4 и 1023 К для SnFe(OH)x ширина дифракционных пиков на половине максимума интенсивности становится меньше, что свидетельствует о росте кристаллитов. Средний размер оксидных кристаллитов, вычисленный по уравнению Дебая-Шерера, растет как с увеличением температуры отжига, так и продолжительности (рис. 3). Допирование железом существенно подавляет рост кристаллитов: размер чистых порошков после отжига при 923 К – около 25 нм, в то время как допированных – около 5 нм, что подтверждают также результаты ПЭМ.
В ряде работ подчеркивается стабильность размеров кристаллитов SnO 2 , полученных методом совместного осаждения солей [31]. Размеры кристаллитов обычно находятся в диапазоне 3-50 нм и слабо изменяются во время термообработки. Методом соосаждения α-оловянной кислоты и гидроксидов металлов раствором аммиака с последующим термическим разложением возможно получение высокооднородных сложных оксидных систем SnO 2 -M n O m (M=V, Fe, Mo, W) [34].
Нанопорошок SnO2, допированный титаном, готовили золь-гель-методом, также используя тетрахлорид олова в качестве исходной соли [32]. SnCl 4 ⋅ 5H 2 O растворяли в смеси этанола и воды (1:1), добавляли тетрахлорид титана (Ti:Sn=5:95) с полиэтиленгликолем (ПЭГ) в качестве диспергирующего агента и перемешивали 30 мин при 353 К. После этого добавляли гидрат аммония до рН=7, выдерживали 48 ч, осадок отделяли центрифугированием, промывали, сушили, отжигали 2 ч при 623, 773 и 973 К. В результате получили нанопорошок с размерами – 191 –
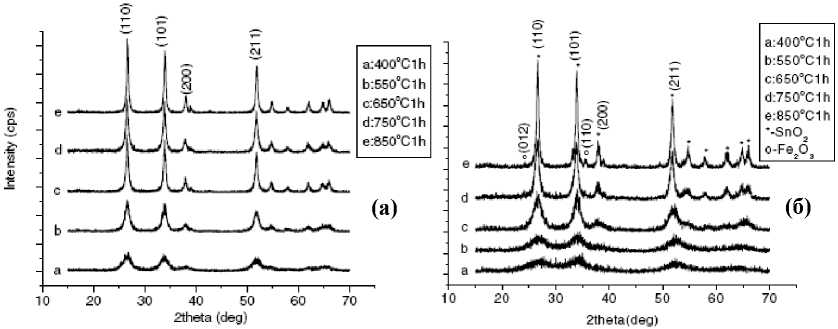
Рис. 2. Влияние температуры отжига на вид рентгенограмм: (а) – Sn(OH) 4 ; (б) – SnFe(OH) x ,
(Sn:Fe = 14:5) [17]
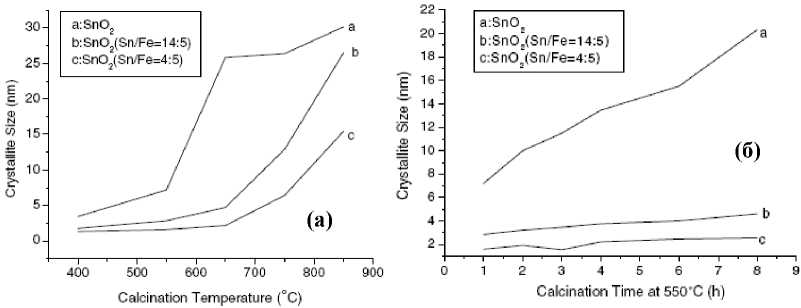
Рис. 3. Влияние температуры (а) и времени отжига (б) на размер кристаллитов в изученных системах [17]
частиц соответственно около 10-20, 30-50 и 70-90 нм, причем присутствие ПЭГ предотвращало агломерацию частиц.
Полученные порошки прессовали, спекали при температуре от 873 до 1473 К и измеряли плотность и электропроводность. Максимальные значения плотности и электропроводности получили при 1273 К: около 6,5 г/см3 и 0,016 Ом-1 ⋅ см-1 соответственно. Для сравнения аналогичным путем получали материал из недопированного SnO 2 . Его плотность и проводимость были значительно ниже: 6,053 г/см3 и 1,84 ⋅ 10-6 Ом-1 ⋅ см-1.
Сравнивая результаты работ [17] и [32], использующих один и тот же прекурсор, но разные методы синтеза гидроксида олова, следует признать метод соосаждения [17] более эффективным как по продолжительности процесса, так и по дисперсности продукта. Кроме того, использование ПЭГ может создать в ряде случаев проблемы применения такого порошка из-за возможности образования коксового остатка при термообработке образцов в инертной среде.
Такое заключение подтверждается данными работы [33], где методом соосаждения из растворов хлоридов SnCl 4 и VCl 3 синтезировали нанопорошки SnO 2 , допированные ванадием (0,10,5 масс. %). Осаждение проводили при 333 К раствором гидроксида натрия. С ростом добавки ванадия средний размер кристаллитов несколько снижается (7,7-5,4 нм). После отжига в воз- – 192 –
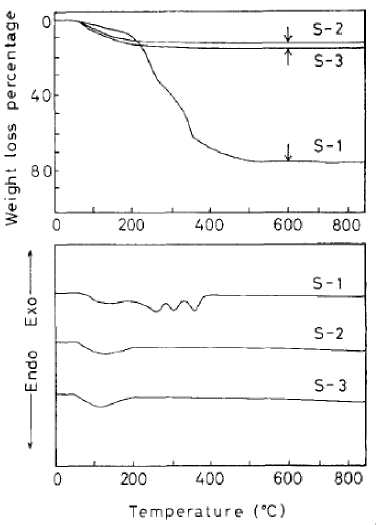
Рис. 4. ТГ-ДТА кривые гидратированных порошков диоксида олова [34] на воздухе, скорость нагрева 2 К/мин духе при 873 К в течение 2 ч наночастицы имели сферическую форму и узкое распределение по размерам (в основном, менее 20 нм).
Интересные экспериментальные результаты сообщены авторами [34, 35]. Синтез гидратированного SnO 2 производили из водного раствора 0,01 М SnCl 4 ⋅ 5Н 2 О и 1 М мочевины при исходном рН=0,5 (добавлена НСl) при температуре 358 К в течение 4 ч (образец S-1). Другие эксперименты были проведены в присутствии ионов SO 4 2- (добавляли 0,015M (NH 4 ) 2 SO 4 +0,008M H2SO4, образец S-2), а также SO42- и SbCl3 (добавляли 0,015M (NH4)2SO4+0,008M H2SO4+0,001M SbCl 3 , образец S-3). Результаты термического анализа образцов существенно различались (рис. 4): образец S-1 имел потерю массы при нагревании в виде воды в интервале температур 473-673 К около 75 %, у S-2 и S-3 – 13,4-15 %, наблюдающуюся в интервале 333-473 К, то есть гидратированный осадок SnO2, полученный без добавления сульфат-ионов был обводнен значительно сильнее, а ДТА-кривая показывала поэтапную дегидратацию связанной воды, в то время как на других осадках связанная вода отсутствовала. Авторы это явление не объясняют. Результаты рентгено-дифракционных измерений практически одинаковы.
Сферические частицы гидратированного SnO2 и частицы в виде стержней в узком диапазоне распределения по размерам получали нагреванием подкисленного раствора SnCl 4 при 373 К в присутствии формамида [36]. Форма частиц зависела от рН суспензии. Гидрогель SnO2 получали также добавлением концентрированного NH 3 ⋅ H 2 O к водному раствору SnCl 4 при рН=1,5 или рН=11 [37]. Количество остаточных ионов Сl– оказывало влияние на равновесие золь-гель в этой коллоидной системе: при повышенных концентрациях гелеобразование затормаживалось.
В последние десятилетия золь-гель-метод привлекает внимание исследователей синтеза оксидов вследствие ряда достоинств: (1) температура синтеза относительно низкая; (2) оксид-– 193 – продукт свободен от посторонних ионов; (3) возможность точного контроля уровня допирования оксида; (4) возможность управления дисперсионным составом в широком диапазоне размеров вплоть до единиц нанометров, а также и морфологией частиц.
Диоксид олова, допированный медью, синтезирован с использованием золь-гель-процесса [38]. Для приготовления растворов водного коллоида в диапазоне концентрации меди 0,7-5,0 М исходными реагентами служили SnCl 4 ⋅ 5H 2 O и CuCO 3 ⋅ Cu(OH) 2 . Осадитель – гидрат аммиака (pH=11). Смесь помещали в тефлоновый контейнер, содержащий жидкий парафин и хлороформ. Ввиду низкой плотности гидрозоль находился на поверхности жидкости. Золь-гель-переход наблюдается при критической концентрации SnO2, равной 0,42 моль/л, и величине парциального давления пара воды, соответствующей температуре 318 К. В итоге термообработки в течение 30 мин при температурах 383, 573 и 873 К получен прозрачный, однородный, хрупкий и микропористый ксерогель. По результатам ТГА отмечается, что количество адсорбированной воды растет с концентрацией меди.
Частицы гидроксида олова размером 3-5 нм синтезировали золь-гель-методом из изопро-поксида олова (IV) Sn[OCH(CH3)2]4: в раствор изопропоксида в изопропаноле добавляли воду и нагревали при 373 К и рН=5,5 в течение 3 ч [39].
В этой же работе изучили золи и ксерозоли, полученные гидролизом 0,384 М раствора SnCl 4 при комнатной температуре в течение 10 лет, а также гидролизом раствора в автоклаве при 433 К в течение 2-24 ч.
Целью работы было выявление различий примененных методов гидролиза Sn4+. Особое внимание было уделено определению размеров частиц методом низкочастотного комбинационного рассеяния. Размер кристаллитов определяли также методом Шерера. Отожженный при 673 К осадок представлял собой частицы, состоящие из нескольких кристаллитов.
Модифицированный золь-гель-метод синтеза SnO 2 с использованием смеси нормального пропанола и изопропанола применен авторами [18]: тетрахлорид олова растворяли в пропаноле (мольное соотношение 1:12) и одновременно готовили раствор изопропанола и воды (мольное соотношение 2:3). Далее второй раствор медленно добавляли к первому (смеси указанных спиртов находились в соотношении 1:2 и 2:1). Полученный осадок гидроксида отжигали при 873 и 1473 К и исследовали рентгеновской дифракцией, ПЭМ, ИК-спектроскопией, а также методом БЭТ определяли удельную поверхность. Изменяя содержание спиртов в растворе, можно в некоторой степени регулировать величину кристаллитов осадка (6 и 10 нм для указанных соотношений и, соответственно, 33 и 23 м2/г), размер которых увеличивается до ~ 80 нм (удельная поверхность 3 м2/г) при температуре отжига 1473 К.
Синтез нанопорошков SnO 2 путем гидролиза хлорида олова (II) в водном растворе при взаимодействии с гидратом аммиака подробно исследовали в работе [15]. Результаты потенциометрического и кондуктометрического титрования позволили рассмотреть механизм процесса и в совокупности с РФА определить оптимальные значения рН=4,6-6,25 (концентрация SnCl 2 =0,3 моль/л), при которых осадок представляет собой простую фазу – касситерит, хотя и не очень хорошо окристаллизованный. Поэтому авторы полагают, что полностью исключать присутствие хлоридов или основных оксихлоридов олова в аморфном состоянии в составе осадка нельзя. Осаждение при рН=10,6 дает четкую дифрактограмму осадка в виде Sn6O4(OH)4. Данные СЭМ и ПЭМ показывают, что после отжига при температурах до 873 К продукт осаж- – 194 –
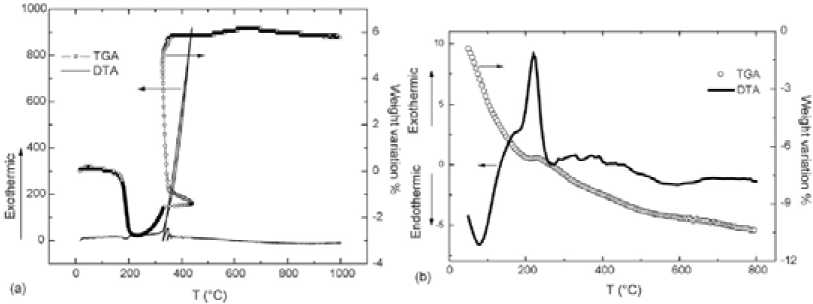
Рис. 5. Термический анализ осадка, полученного из 0,3 моль/л раствора SnCl2 при рН=6,25: (а) - осадок до отмывки, (b) – отмытый (условия: поток воздуха, скорость 5 град/мин) [15]
дения представляет собой губчатые частицы размером около 5 мкм, состоящие из кристаллитов 20-60 нм, с удельной поверхностью около 19,5 м2/г.
Интересны данные термического анализа (ТГ и ДТА) в этой системе, существенно зависящие от присутствия Сl-ионов: кривые на рис. 5а принадлежат осадку до отмывки его от Сl–, на рис. 5b - отмытому с помощью раствора диэтиламина продукту. Резкий прирост массы образца и соответствующий значительный тепловой эффект при температуре около 573-623 К (300350 °С) (рис. 5а) вызваны, по мнению авторов, окислением Sn+2, присутствующим в образце в виде аморфного SnO. Кристаллизация касситерита и фазовые переходы соединений в SnO2 также дают вклад в этот экзотермический пик около 623 К (350 °С). Все процессы заканчиваются при температуре всего около 653 К (380 °С), и далее изменения массы практически не наблюдается. Этот нетривиальный факт авторы не обсуждают, он нуждается в дополнительном изучении и объяснении. Вызывает также вопросы значительный прирост массы (около 9 %), что могло бы наблюдаться при окислении образца, состоящего практически из чистого монооксида SnO. В таком случае было бы более понятно и постоянство массы в интервале 653-1273 К (380-1000 °С).
Кривые отмытого осадка (рис. 5b) имеют обычный вид: медленная потеря массы вплоть до максимальной температуры эксперимента 1073 К, что характерно и для других аналогичных систем, в соответствии с данными ряда авторов. Эндоэффект обусловлен выделением воды, а экзоэффект (около 493 К) – кристаллизационными процессами.
Простой в реализации метод синтеза наносферического порошка SnO2 предложили авторы работы [40]: порошок металлического олова окисляли в растворе пероксида водорода в присутствии поливинилпирролидона (ПВП) и этилендиамина (ЭДА). Полученную суспензию промывали водой и сушили на воздухе. Рентгенограммы такого осадка показывали структуру рутила и средний размер кристаллитов от 2,8 до 9,5 нм в зависимости от содержания ПВП. ПЭМ и СЭМ-микроскопия показали практически монодисперсную сферическую морфологию осадка с размером частиц около 30 нм, которые, в свою очередь, состояли из кристаллов величиной порядка 4 нм.
Вторичные сферические частицы образуются только в присутствии ПВП, а их размер зависит от концентрации ПВП в растворе: с ростом концентрации он уменьшается. В отсутствие ПВП формируются агрегаты нерегулярной формы из кристаллитов величиной около 2,5 нм.
Отжиг в течение 4 ч при 573 К укрупнил наносферы до 40-80 нм и вызвал появление агрегатов (порядка 200 нм), т.е. ПВП при этой температуре уже теряет защитные свойства от агрегации и спекания частиц. ЭДА действует в рассматриваемом процессе как катализатор, радикально ускоряя окисление олова. Такой же эффект наблюдали и при синтезе наночастиц Sb2O3 из металлической сурьмы [34].
Допированный сурьмой диоксид олова используют для органических светоизлучающих диодов. Проводящие пленки, толщина которых около 100 нм [41], наносят на прозрачную непроводящую подложку, используя золь-гель-метод. Технология предполагает 4 стадии: гидролиз, поликонденсацию, сушку и уплотнение. После осаждения пленку просушивают 40 мин при температуре 423 К и затем отжигают при 773 К. Данный метод в измененной форме может быть использован для получения осадков, состоящих из смеси оксидов сложной структуры, если вместо подложки использовать, например, ионы металла или молекулы оксида металла и на них из раствора осаждать гидроксиды другого металла.
В работе [20] проведены исследования по получению осадков в системе «Sn(n)-Sb(III)-H2O» из водных растворов SnCl2 и SbCl3 с целью изучения процессов фазообразования, протекающих при совместном осаждении и последующей термообработке. В качестве осадителя использовали гексаметилентетрамин (ГМТА). Полученные осадки исследованы с привлечением ИК-спектроскопии, рентгенофазового и термогравиметрического анализа. На пленках изучены электропроводящие свойства. С целью контроля процесса осаждения в систему вводили ионы фтора для образования устойчивых комплексов. Согласно исследованиям авторов, присутствие иона фтора значительно влияет на процесс осаждения. Графические зависимости представлены на рис. 6.
Растворы хлорида олова и хлорида сурьмы устойчивы только при добавлении в систему соляной кислоты. Этот факт также отмечается и в других работах [42, 43]. Осадок такого же состава «Sn(II)-Sb(III)-H2O» получали методом термического разложения смеси гидроксидов при температуре 1270 К в течение 2 ч [42]. Другие авторы [43] целевой продукт состава SnO2-Sb получали методом совместного осаждения солей. Осажденную смесь подвергали сушке при 318 К (48 ч) и последующему отжигу при температуре 1473 К (7 ч).
Методом соосаждения из растворов солей SnCl4 и Ce(NO3)3 в этаноле в присутствии ПАВ (бромид цетилтриметиламмония) при рН=7 получали осадок гидроксидов добавлением гидрата аммония [44]. Осадок затем подвергали старению при 300 К в течение 24 ч для получения допированного SnO2 и далее отжигали 2 ч на воздухе при 773, 873, 973 и 1073 К. Ограненные кристаллы SnO2 размером 10-16 нм наблюдаются на ПЭМ-снимках, что свидетельствует об эффективности примененного ПАВ, предотвращающего агрегацию даже после отжига при 773 К. После отжига 973 и 1073 К частицы увеличиваются до 25-27 нм, в то время как удельная поверхность уменьшается от максимального значения 60 до 34 м2/г.
В работе [45] применили следующую методику осаждения. Раствор SnCl2 (0,1 моль/л) предварительно гидролизовали в течение 144 ч с обратным холодильником и затем нейтрализовали гидратом аммония. Осадок промыли, высушили и отожгли при 873 и 1173 К. ПЭМ-снимки показали, что продукт представляет собой рыхлые частицы-агломераты нерегулярной формы, состоящие из кристаллитов размером до 20 нм.
Подробное сравнительное изучение двух экспериментальных методов синтеза диоксида олова проведено авторами [46]: окислением порошка олова в HNO 3 (с целью исключения хлора – 196 –
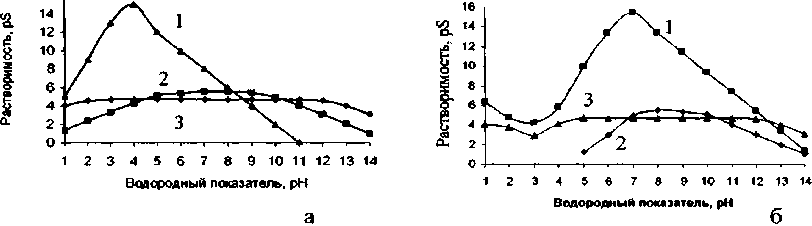
Рис. 6. Зависимости растворимости гидратированных оксидов металлов в водных (а) и фторидных растворах, при cF=0,1 мол/л: 1 - Sn(IV), 2 - Sn (II), 3 - Sb (III) [20]

Рис. 7. СЭМ-снимки отожженных осадков [46]
из процесса синтеза и твердого продукта) и осаждением из раствора тетрахлорида олова при помощи гидразина. Окисление порошка олова проводили в растворе азотной кислоты концентрацией 34 об. %. Во втором методе 0,6 М водный раствор SnCLp5H2O (рН = 1,0) медленно добавляли к перемешиваемому раствору гидразина (26 об. %, рН = 11,0) при комнатной температуре и выдерживали, перемешивая, в течение 10 дней. Уменьшение этого времени созревания вело к уменьшению удельной площади отожженного при 873 К осадка. В обоих методах осадок центрифугировали, несколько раз промывали водой, сушили, отжигали. Выход в первом методе составил 85 %, во втором – 75 %. Несмотря на тщательную отмывку, отожженный при 873 К ксерогель, полученный из хлорида, содержал 400 ppm хлора. На всех этапах обработки, от свежеосажденных образцов до отожженных, ксерогели показывали структуру касситерита с постепенным сужением основных дифракционных пиков при повышении температуры термообработки. Морфология частиц существенно различалась (рис. 7), хотя оба отожженных осадка имели весьма широкое распределение по размерам.
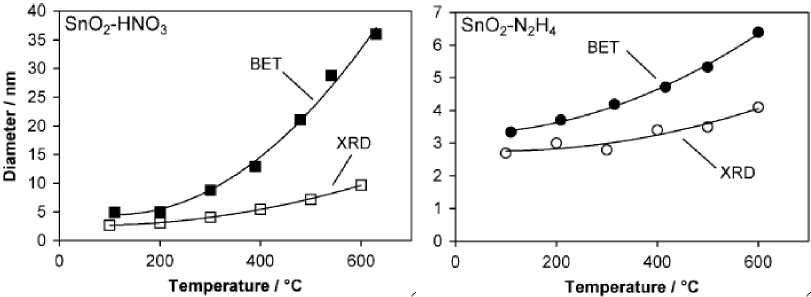
Рис. 8. Изменение размеров кристаллитов (рентгеновская дифракция) и размеров частиц (метод БЭТ) в зависимости от температуры обработки [46]
Крупные частицы представляли собой агрегаты из более мелких частиц диаметром в несколько десятков нанометров, пронизанных порами размером от единиц до десятков нм. В соответствии с этим осадки имели высокую удельную поверхность около 100-250 м2/г (определено методом БЭТ).
Диаметр кристаллитов, определенный из рентгеновской дифракции и рассчитанный из БЭТ-измерений в предположении сферической формы частиц, зависел от температуры термообработки (рис. 8).
Эти же осадки подробно изучены ультрафиолетовой и инфракрасной спектроскопией с целью исследования нарушений кислородной стехиометрии и выявления процессов дегидроксилирования поверхности. Показано, что метод получения влияет не только на морфологию осадка, но и на его полупроводниковые свойства: например, с повышением температуры обработки заметно снижается кажущаяся величина запрещенной зоны.
При работе с гидроксидами олова, как правило, возникает проблема присутствия нежелательных ионов в растворе. Аморфные осадки a-SnO2 - nH2O, полученные из хлорсодержащих прекурсоров, требуют обычно дальнейшей тщательной отмывки от Cl-, а кроме того, от ионов щелочных металлов, например Na + . Присутствие посторонних ионов влияет не только на свойства конечного продукта, но и на сам процесс осаждения [15, 20]. Водные растворы H2[SnCI 6 ] можно нейтрализовать любыми щелочными реагентами, однако с учетом высокой сорбционной способности продукта предпочтительным осадителем является раствор аммиака, что подтверждается использованием его в ряде работ [15, 17, 31, 37, 38, 44, 45].
Метод соосаждения солей металлов и оксидных смесей находит, по-видимому, наибольшее применение. Его основное преимущество заключается в возможности широкого варьирования параметров процесса, в частности, рН раствора. С ростом величины рН раствора степень гидролиза растворенных форм увеличивается, протекают процессы поликонденсации, приводящие к образованию золей и гелей гидроксидов (золь-гель-метод). Эти частицы способны сорбировать из раствора ионы за счёт поверхностных гидроксогрупп. В процессе нейтрализации кислых растворов соединений Sn(IV) (независимо от способа нейтрализации) образуется не только SnO2-nH2O, но и полимерные основные соли. При этом рН осаждения гидроксида олова (IV) может изменяться в широких пределах (от 1,2 до 5,1) в зависимости от природы пре-– 198 – курсора, его концентрации в растворе, температуры, природы растворителя, концентрации и природы примесей. Изменение условий осаждения фазы влечет изменение её состава и строения. Если гидролиз соединений олова (IV) проводится при температурах не выше 290 К, то осаждается гидрооксид α-SnО2⋅nH2О, растворяющийся в кислотах и щелочах. При повышенной температуре из растворов осаждается β-форма SnО2⋅nH2О, которая химически инертна [47-49].
Наряду с использованием неорганических прекурсоров широко применяется синтез SnO 2 из солей органических кислот: изопропоксид олова (IV) Sn[OCH(CH3)2]4 [39, 50], тартрат олова (виннокислое олово) [51], оксалат олова SnC 2 O 4 и др. Рассмотрение методов с использованием этих и некоторых других соединений изложено ниже.
Получение SnO2 из солей органических кислот
Анализ литературы показывает, что в схемах синтеза диоксида олова преимущественно нашли применение соли щавелевой и лимонной кислот. Наиболее часто в работах фигурирует оксалат олова (II) SnC 2 O 4 благодаря своим свойствам [52]: аморфное вещество, не растворяющееся в воде с температурой разложения около 553 К. Эту соль используют как в виде готового реактива, так и в качестве промежуточного продукта в технологической схеме синтеза SnO 2 .
В ряде работ [14, 23, 53-55] диоксид в нанодисперсной и субмикронной форме получали термическим разложением оксалата, осажденного из смеси растворов хлорида олова (II) и щавелевой кислоты при комнатной температуре. Следует сказать, что такой способ синтеза SnC 2 O 4 как прекурсора для дальнейшего получения наноразмерных форм SnO 2 путем термического разложения, пожалуй, наиболее распространен в современных работах и еще будет упоминаться ниже. Полученный фильтрованием осадок промывали, сушили, отжигали при 1073 К в течение часа на воздухе и затем растирали. Независимо от концентрации реагентов (0,04-0,2 моль/л) формировались удлиненные призматические кристаллы оксалата, которые превращались после термического разложения в сферические кристаллы тетрагонального SnO 2 размером около 75 нм (оценивались электронной микроскопией и лазерной дифракцией). Последние агломерировались в пластинки диаметром порядка 0,5 мкм, морфологию которых связывают с пластической деформацией при растирании отожженного продукта. Считают также, что этому способствует адсорбированная на поверхности порошка атмосферная влага.
В тщательно и подробно проведенной материаловедческой работе [24] авторы применили термический и химический метод синтеза диоксида из оксалата. Термический анализ процесса разложения готового порошка реактива SnC2O4 в статической воздушной атмосфере показал потерю массы около 30 % в узком интервале температур вблизи 623 К, величина которой согласуется с теоретическим значением 27,1 % в соответствии с общим уравнением разложения
SnC 2 O 4 = SnO 2 + 2CO.
Как показывают ПЭМ-снимки, частицы SnO 2 , полученные этим методом (рис. 9), сохраняют размеры и форму частиц прекурсора. Однако текстура этих частиц микронного размера характеризуется наноразмерными (около 50-70 нм) ограненными областями и наличием значительной пористости. Рентгенофазовый анализ таких образцов, отожженных при 723 К в течение различных периодов времени, показывает структуру рутила. Из этого следует, что – 199 –

Рис. 9. ПЭМ-снимки образцов SnO2, синтезированных термическим разложением оксалата при 723 К (а) и химическим окислением SnC2O4 с последующим отжигом при 723 К (б); длина полосы увеличения: (а) – 150 нм, (б) – 50 нм [24]
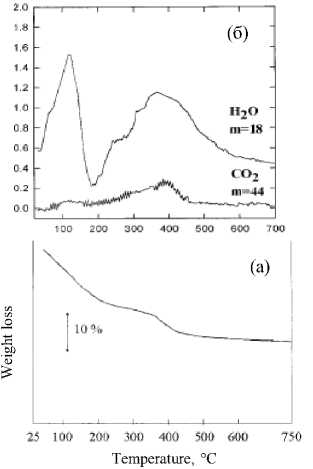
Рис. 10. ТГ-кривая (а), кривые интенсивности выделяющихся газов (б) и рентгенограммы после соответствующих термообработок осадка на воздухе в течение 12 ч (в); здесь нижняя кривая – для просушенного осадка [24]
(в)
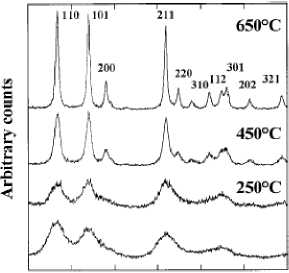
20 ЛО 40 50 60 70 NO
Angle (2 theta)
из микроразмерных частиц соли в процессе ее термического разложения формируются наночастицы диоксида, агломерированные в пористый агрегат, по форме и размеру повторяющий исходную частицу оксалата.
Получение диоксида химическим методом авторы проводили растворением порошка SnC 2 O 4 в 33 %-ном пероксиде водорода при комнатной температуре. Затем прозрачный раствор медленно нагревали до кипения при перемешивании. Выпаривание воды приводило к получению белого коллоидного раствора и далее – твердого осадка, который сушили при 333 К, подвергали термической обработке и тестированию дифракционными, спектральными методами, микроскопией и термическим анализом.
Термогравиметрический анализ просушенного осадка проводили в динамической среде азота с одновременной масс-спектрометрией выделяющихся газов (рис. 10 а, б). Очевидно, что – 200 – вода удаляется из образца в два основных этапа: ниже 473 К - несвязанная и выше этой температуры, вплоть до 973 К – структурная вода.
Рентгенограммы (рис. 10 в) исходного просушенного осадка, а также термообработанного на воздухе в течение 12 ч при указанных на рисунке температурах, существенно изменяются и выше 723 К принимают вид, характерный для рутила SnO2. Авторы полагают, ссылаясь на литературные данные, что уширенные пики при низких температурах связаны с тенденцией высокодисперсного диоксида олова к кристаллизации в орторомбической структуре. ПЭМ-наблюдения как будто бы подтверждают это: просушенный осадок представляет собой нерегулярные агломераты ультрадисперсных частиц со средним размером около 3 нм, который увеличивается с температурой отжига в ряду 523, 723, 923 и 1073 К соответственно до 5, 7, 12 и 16 нм (рис. 9 б). Ультрадисперсная структура просушенных образцов хорошо согласуется с данными рамановской спектроскопии.
Более вероятным объяснением уширения дифракционных пиков является слабая окри-сталлизованность образца, что может быть связано с образованием осадка в виде α-оловянной кислоты SnO2 - nH2O. Как известно [30], свежеосажденная кислота рентгеноаморфна, но при старении и удалении воды на рентгенограммах все более проявляются линии, соответствующие кристаллической SnO2. Такое объяснение сочетается с плавно ниспадающим видом ТГ-кривой (рис. 10а) и растянутыми по времени тепловыми эффектами выделения воды.
В работах [26, 56] авторы также синтезировали диоксид разложением оксалата, но прекурсор получали дозированным смешением растворов хлорида SnCl2 - 2H2O (0,5 моль/л) и осадителя - щавелевой кислоты (COOH)2 - 2H2O (0,5 моль/л) при повышенных температурах (303-363 К). Далее белый осадок отделяли, промывали и сушили при 363 К в течение 24 ч. Разложение проводили при температуре 673-773 К с заданной скоростью нагрева (1 и 3,3 К/с [56] и от 4,2 до 100 К/с в микроволновой печи [26]).
Термический анализ просушенного осадка на воздухе показал, что в интервале температур от комнатной до 673 К резкое уменьшение массы образца в результате термолиза наблюдается от ~543 до 643 К и сопровождается широким экзотермическим пиком на ДСК-кривой с максимумом около 593 К. В этот интервал укладываются значения температуры разложения, приведенные в работе [24] (623 К), а также в [52] (553 К).
Рост температуры в процессе осаждения приводит к формированию осадка в виде усов диаметром 0,5-2 мкм и длиной 40-100 мкм, в то время как при 303 К морфология осадка осколочная с весьма широким распределением по размерам от долей до десятков мкм. Как и в работе [24], на основании данных просвечивающей электронной микроскопии отмечено, что после термолиза соли первичная форма частиц остается прежней, но они уже представляют собой пористые агрегаты, образованные сферическими наночастицами. Причем влияния скорости нагрева при термолизе на размеры наночастиц не замечено: во всех случаях этот размер составляет около 20-30 нм (т.е. заметно меньше, чем в [24], что говорит о влиянии предыстории исходной соли), а удельная площадь поверхности, определенная методом БЭТ, находится в пределах 21-24 м2/г.
В ряде работ решали более сложную задачу синтеза диоксида олова, допированного различными примесями [8, 21, 50, 57, 58]. Авторы [58] использовали простой метод сухого смешения товарных солевых прекурсоров с последующей термообработкой: SnC2O4, Ce(SO4)2-4H2O – 201 – и (NH4)2Ce(NO3)6. Исходные соли и их смеси подвергали термическому и рентгенофазовому анализу. ТГ- и ДТА-кривые оксалата, снятые в статической атмосфере воздуха до 1273 К при скорости нагрева 10 К/мин показывают узкий интервал разложения около 623-653 К и суммарное изменение массы 33,66 %, что заметно превышает приведенную выше расчетную величину 27,1 % (это различие авторы не обсуждают). Продукт, по данным РФА - SnO2. В то же время, нагрев до температуры 773 К как чистого оксалата, так и смеси оксалата и соли церия в соотношении 39:1 приводит к образованию SnO2 и некоторого регистрируемого количества SnO.
Высокодисперсный диоксид, допированный железом, кобальтом, ниобием и лантаном, синтезировали методом полимерных прекурсоров (метод «Pechini», цитратный метод), использовав в качестве источника олова оксалат или дихлорид олова (II). Для сравнения применили также традиционный метод мокрого (изопропанол) смешения оксидов в высокоскоростной турбине, что обеспечило получение порошка с удельной поверхностью 7,2 м2/г и средним размером частиц ниже 300 нм. Конечной целью было получение компактного спеченного материала для варисторов [8, 21].
Практически процесс осуществляли следующим образом [21]. Раствор, содержащий катион Sn +4 , получали приливанием азотной кислоты в дисперсию SnC2O 4 в воде при постоянном перемешивании. Затем добавили раствор лимонной кислоты в этиленгликоле (молярное соотношение 1:4) и, перемешивая, выдерживали 24 ч для образования цитратов. Прозрачный раствор далее нагревали до формирования аморфной смолы, которую прокаливали при 623 К в течение 12 ч для термолиза органической составляющей. Полученную черную хрупкую массу перетирали, обжигали в муфельной печи при 873 К в течение 10 ч, прессовали в виде дисков и спекали 2 ч при 1573 К. После обжига при 873 К РФА показал присутствие только кристаллического SnO2 в нанодисперсном виде со средним размером частиц 15-20 нм и удельной поверхностью 30 м2/г.
С дихлоридом олова (II) как прекурсором вместо SnC2O4 [8] процесс на первых этапах имел отличия: из кислого (HNO 3 ) раствора дихлорида олова (II) добавлением гидрата аммиака осаждали диоксид олова, который далее тщательно промывали от Cl-ионов и вместе с требуемым количеством допирующих добавок сливали с раствором лимонной кислоты в этиленгликоле. Дальнейшие процедуры аналогичны указанным выше. Полученный таким способом порошок имел удельную поверхность 9 м2/г, широкое распределение по размерам и был агломерирован в слабосвязанные агрегаты, которые легко разрушались при размоле. После размола удельная поверхность достигала 25 м2/г, а распределение по размерам частиц становилось более однородным.
Используя этот же «полиол-метод» с лимонной кислотой в качестве хелатирующего агента и этиленгликолем как полиол-растворителем, авторы [13] получали допированные сурьмой (0, 6, 10, 14 мол. % Sb) наноразмерные порошки диоксида олова. Порошки отжигали при 1073 и 1473 К и характеризовали с помощью методов рентгеновской дифракции. Во всех случаях фиксировали только SnO2-фазы рутила, а сурьма, замещающая Sn + 4 в узлах решетки, присутствовала в виде Sb + 3 и Sb + 5 в зависимости от условий. С ростом температуры отжига, как и следовало ожидать, наблюдается укрупнение частиц, допирование снижает этот эффект.
Ультрадисперсный SnO2 с кристаллитами менее 5 нм получен [59] путем нагревания дихлорида SnCl2-2H2O (11,28 г) в этиленгликоле (100 мл) при 468 К на масляной бане в течение 4 ч – 202 – с энергичным перемешиванием. Охлажденный коллоидный раствор разбавляли ацетоном для удаления избытка этиленгликоля, центрифугировали и осадок сушили в вакууме при 353 К. Осадок имел удельную поверхность 127 м2/г со средним размером зерна, согласно прямым измерениям по ПЭМ-снимкам, около 4,7 нм.
Нагревание смеси оксалата олова (II) и поливинилпирролидона (ПВП) в этиленгликоле в течение 3 ч при 468 К приводило к формированию нанопроволок со средним диаметром 50 нм и длиной до 30 мкм. После отжига при 773 К каждая нанопроволочка превратилась в высокопористую структуру, состоящую из нанокристаллитов SnO2 размером около 5 нм [60].
Аналогичные процедуры при отсутствии ПВП в растворе приводят к получению поли-кристаллических нанопроволок SnO2 диаметром 250-800 нм в зависимости от концентрации оксалата [61]. При этом размер первичных зерен составляет около 50 нм.
Таким образом, в рассмотренных работах [8, 21, 59-63] прекурсорами диоксида олова в действительности служат цитраты, которые довольно широко используются для синтеза сложных оксидных фаз в различных методических модификациях цитратного способа (см., например, [30]). Применяя термическое разложение цитрат-геля олова [22], авторы получали и исследовали нанопорошки SnO2. Комплекс цитрата олова Sn3C12H10O14 готовили растворением металлического олова в соляной кислоте, добавляя затем лимонную кислоту Состав подтвердили химическим анализом, ЯМР- и ИК-спектроскопией. Термический анализ (ТГ и ДТА) показал, что полное разложение прекурсора оканчивается около 773 К. Прокаливание комплекса при температуре 773 К и выше приводит к синтезу кристаллического SnO2 тетрагональной (рутильной) структуры. Кристалличность увеличивается с ростом температуры прокалки и вместе с тем растет средний размер наночастиц: 873 К (17 нм), 973 К (25 нм), 1173 К (41 нм), 1273 К (68 нм).
Микропорошки размером около 8-15 мкм системы TiO2-SnO2, богатых титаном составов (1-x)TiO2-xSnO2, x=0,05-0,3, получали сжиганием смеси оксалата олова с изопропоксидом титана (IV) Ti[OCH(CH3)2] 4 [50]. Как топливо использовали мочевину.
Представляет интерес, с точки зрения расширения номенклатуры прекурсоров для синтеза наноформ SnO2, работа [25], в которой авторы синтезировали и исследовали более сложную соль Sn2(NH 4 )2(C2O 4 )3 - 3H2O. Ее получали путем добавления оксалата олова в раствор оксалата аммония (NH 4 )2C2O 4 - H2O при температуре 338 К и перемешивании. Последующее выпаривание раствора при комнатной температуре приводило к выделению прозрачных призматических кристаллов целевой соли совместно с (NH 4 )2C2O 4 - H2O. Чистого аммонийного оксалата олова выделить не удалось. Анализ термогравиметрической кривой полученной смеси (рис. 11) показывает, что в процессе нагревания при температуре около 470 К образуется оксалат олова (II), который далее в интервале температур 500-540 К переходит в диоксид. Высокотемпературный РФА подтвердил эти результаты.
Таким образом, термолиз оксалата в данном случае наблюдается при значительно более низкой температуре, чем во всех случаях, приведенных выше. Это, по-видимому, связано с повышенной реакционной способностью и высокой дисперсностью SnC2O 4 , являющегося здесь промежуточным продуктом твердофазного превращения. Различие составляет около 100 К, что может быть весьма существенным, например, с точки зрения стабильности синтезируемых нанопорошков: пониженная температура синтеза способствует сохранению размеров частиц – 203 –
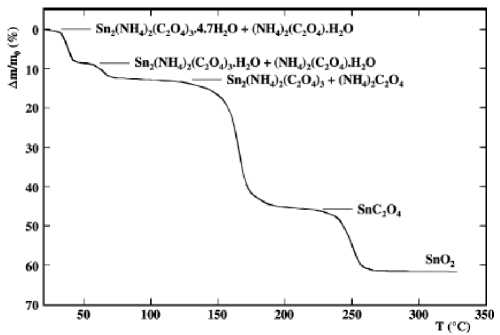
Рис. 11. Термогравиметрическая кривая разложения смеси солей в воздухе (10 К/мин) [25]
и предотвращает агрегирование. Кроме того, в конкретном процессе может быть полезной повышенная реакционная активность в твердофазных взаимодействиях синтезируемого при пониженных температурах SnO2.
Несмотря на широкую распространенность метода термического разложения, качество материала, получаемого этим способом, все же уступает качеству материала, приготовленного из полимерных прекурсоров. Хотя метод полимерных прекурсоров требует значительных временных затрат, он обеспечивает гомогенное распределение допирующих добавок. Процесс, как следует из изложенного материала, включает использование растворимых солей представляющих интерес металлов и базируется на способности а-гидроксикарбоновых кислот образовывать хелаты. При нагревании в присутствии полигидроксиспиртов эти хелаты подвергаются полиэтерификации, и катионы оказываются однородно распределенными в результирующей полимерной структуре. Прокаливание полимера приводит к образованию оксидного порошка – высокодисперсного и химически однородного. Кроме высокой эффективности, метод прост и технологичен, использует доступные реактивы, дешев в реализации. Поэтому в последние годы он находит достаточно широкое распространение для получения различных простых и сложных оксидов (ZnO, Al2O3, Bi2O3, Fe2O3, MgAl2O 4 , CoAl2O 4 и др.), в том числе и для синтеза наноформ диоксида олова [8, 21, 59, 60, 61, 63].
Также стоит отметить, что использование оксалата олова, как в качестве прекурсора, так и в виде промежуточного соединения, наиболее эффективно в процессах осаждения из растворов. Установлено [24], что размер и форма частиц осадка оксалата зависит от температуры осаждения, а термообработка просушенного осадка приводит к увеличению агломератов. Однако конечные размеры частиц полученных термическим разложением, значительно больше размеров агломерированных частиц осадка после термического отжига.
Очевидно преимущество методов, включающих в себя стадию проведения синтеза в жидкой фазе, так как предоставляется возможность управлять морфологией и дисперсностью продуктов в широких пределах за счет варьирования параметров. Но, как уже было отмечено выше, существует вероятность роста оксидов при прокаливании. Поэтому целесообразно использовать прекурсоры, позволяющие получать конечные соединения, увеличение кристаллитов которых незначительно. Так, например, в работе [64] исходным реагентом выступает – 204 – сульфат олова (II) (0,20 М), окисленный пероксидом водорода. Размеры кристаллов SnO2 после рекристаллизации увеличились незначительно – с 4-5 до 5-7 нм.
Получение SnO2 разложением сульфата олова (II)
Хорошо известно и подтверждается изложенным выше экспериментальным материалом, что характерные температуры протекания процессов разложения термически нестабильного соединения зависят от его предыстории, режимов испытаний, а также от характера газовой среды. В связи с этим для сознательного осуществления синтеза порошков оксида желательно изучение процессов разложения используемого прекурсора в конкретных условиях.
Одним из немногих подходящих по своим физико-химическим свойствам соединений олова в применении к синтезу SnO2 в значительных объемах для практических целей, а также наиболее доступных по распространенности и стоимости, является сульфат олова (II). Это стабильное при комнатной температуре, негигроскопичное и не имеющее гидратов соединение, в то же время с относительно высокой растворимостью в воде и термически разлагающееся при не слишком высоких температурах. Как упоминалось выше, проблемами применения соединений олова (II) могут быть: высокий восстановительный потенциал в растворах и возможность образования монооксида SnO при термолизе, особенно в инертной среде. Эти вопросы, требующие специального рассмотрения и анализа в рассматриваемом применении, а также термическое разложение сульфата, экспериментально изучены авторами.
Использовали товарный химический реактив сульфата олова (II) квалификации «чда». Термические исследования проводили на приборе синхронного термического анализа Netzsch STA 449С Jupiter. Проводили ТГ- и ДСК-анализ в проточной атмосфере несущего газа аргона (скорость потока 25 мл/мин) при скоростях нагрева образцов 5 и 10 К/мин в алундовых и платиновых тиглях в диапазоне температур, как правило, от комнатной до 1273 К.
Результаты проведенных измерений процесса термического разложения SnSO 4 представлены на рис. 12. Характер ТГ- и ДСК-кривых свидетельствует, что в заметной степени эта соль начинает разлагаться в твердой фазе при температуре около 623 К без предварительного плавления, а заканчивается этот процесс практически полностью лишь к 970 К. Расчетное изменение массы образца, соответствующее суммарному уравнению разложения с твердым продуктом в виде диоксида олова
SnSO 4 = SnO 2 + SO 2
составляет 29,9 % при экспериментальном значении 28,06 %. Такое совпадение может быть признаноудовлетворительным и однозначно свидетельствует о преимущественном образовании SnO2 в результате термического разложения используемой соли в условиях эксперимента.
Очевидно также, что основное изменение массы происходит в интервале 723-803 К с дальнейшим плавным снижением, сопровождающимся двумя относительно слабыми, широкими эндотермическими пиками сложной формы на ДСК-кривой. Это может быть объяснено физическими изменениями в образце в процессе кристаллизации новых фаз, а также сложным механизмом химических стадий на завершающих этапах разложения.
К практически аналогичным результатам по термическому разложению SnSO4 пришли авторы работы [27], изучавшие данный процесс в среде азота. Они нашли, что основные пре- – 205 –
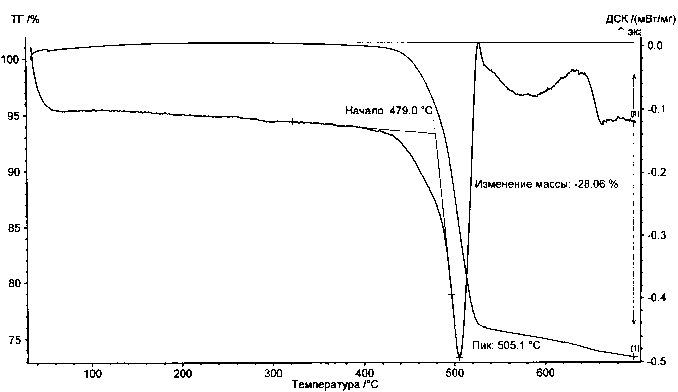
Рис. 12. ТГ- и ДСК-кривые нагревания SnSO4. Тигель – Pt, 10 К/мин, навеска 16,74 мг вращения происходят в интервале температур 723-823 К по приведенному выше суммарному уравнению с образованием диоксида олова и газообразного оксида серы (IV) при измеренной величине изменения массы образца 29,36 %.
Расчетами, основанными на проведенных измерениях, авторы получили S-образную форму кинетических кривых для различных температур процесса и кинетические характеристики реакции разложения. Форма кривых свидетельствует об автокаталитическом характере разложения, а скорость процесса сильно зависит от температуры (значение энергии активации 250,3 кДж/моль). Полное разложение соли происходит менее чем за 20 мин уже при температуре Т ~ 780 К.
Другие исследователи [28] теми же методами изучали термическое разложение метил-сульфоната олова (II), которое протекает в несколько последовательных стадий, одним из его промежуточных продуктов, по их мнению, является сульфат SnSO4. Авторы пришли к выводу, что это соединение формируется при температуре около 663-680 К и далее в узком интервале температур до 698 К разлагается с выделением монооксида SnO. Такие существенные отличия характерных температур по результатам [28] от работы [27] и наших данных могут быть объяснены, по-видимому, высокой реакционной способностью сульфата, являющегося в данном случае промежуточным продуктом кристаллизации на предыдущих стадиях процесса разложения и, скорее всего, находящегося в аморфном или высокодисперсном кристаллическом состоянии.
Утверждение авторов о синтезе SnO при температуре 680-698 К вызывает сомнение, т.к. известно [65, 66], что монооксид устойчив только до температуры около 570 К. Выше он становится неустойчивым и переходит в смесь Sn ( ж ) +SnO2 ( тв ) . В связи с этим важной для анализа взаимодействий в изучаемой системе представляется реакция диспропорционирования монооксида олова в случае его образования при термическом разложении сульфата олова (II), как и других термически нестабильных солей олова (II) в инертной атмосфере.
Расчеты реакции диспропорционирования монооксида олова [67] свидетельствуют о том, что до ~1357 К изменение энергии Гиббса реакции отрицательно и, следовательно, – 206 –
(а)
(б)
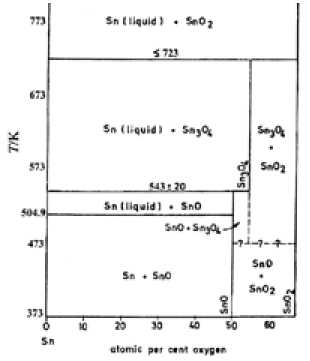
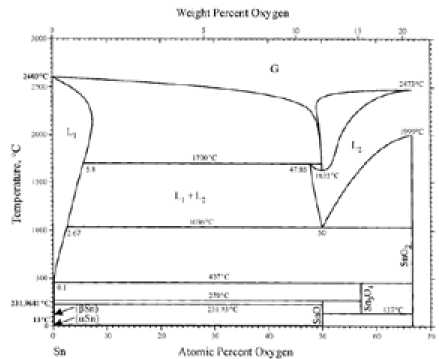
Рис. 13. Фазовая диаграмма Sn-O; а – [1], б – [70]
реакция имеет развитие, а диоксид олова относительно стабилен. Выше этой температуры SnO2 становится менее стабильным, а стабильность монооксида снова возрастает. По другим данным [67], низкотемпературный предел стабильности SnO 658 К. В соответствии с приведенной на рис. 13 фазовой диаграммой эта температура заметно ниже: 543 ± 20 К. Есть также указание на то, что без доступа кислорода SnO диспропорционирует на олово и диоксид уже при температурах выше 453 К [69]. Таким образом, однозначное заключение о температурных пределах стабильности SnO на основании имеющихся литературных данных сделать затруднительно.
На рис. 13 приведены наиболее поздние из опубликованных вариантов фазовой диаграммы системы Sn-O [1, 70]. Результаты экспериментального изучения (рис. 13а) ограничены температурой около 770 К. Полная фазовая диаграмма [70] получена недавно расчетным путем и представлена на рис. 13б в широком интервале температур, вплоть до температуры кипения олова. В этой работе, по-видимому, наиболее полно и подробно проанализированы фазовые термодинамические исследования в системе Sn-O, проведенные за весь предшествующий период. Обе диаграммы практически идентичны в низкотемпературной области и в основном не расходятся с давними оценками работы Платэ и Мейера [65].
Отличие рис. 13а и 13б заключается лишь в небольшой разнице температур (7 К) перитектической реакции Sn + 2SnO2 = Sn3O4.
Более ранняя диаграмма (1949 г), вошедшая в справочники [68, 71], существенно отличается от рис. 13б в низкотемпературной области и, в частности, не содержит равновесий с участием монооксида.
Эти различия свидетельствуют о сложности и кинетических затруднениях взаимодействий, что приводит к неоднозначности в интерпретации результатов фазовых исследований. Большее доверие вызывают диаграммы, результаты которых получены в более поздний период и качественно подтверждаются современными экспериментальными работами, согласно которым монооксид SnO стабилен от комнатных температур до 543 К. Далее он диспропор-ционирует на Sn и Sn3O4, область сосуществования которых доходит до 723 К. Выше ~723 К и – 207 –

Рис. 14. Снимки порошка SnO2, полученного из сульфата олова (II): (а) - *150, (б) - х10000
вплоть до 1309 К стабилен диоксид. При более высоких температурах снова стабилизируется газообразный SnO.
Основываясь на этом выводе, следует заключить, что при разложении прекурсора для SnO2 желательны температуры выше указанного граничного значения 723 К. При более низких температурах в инертной среде может образоваться монооксид олова, одним из продуктов диспропорционирования которого при дальнейшем нагревании является металлическое олово.
Эксперимент по синтезу порошка диоксида олова из SnSO4 без использования стабилизаторов был проведен в печи сопротивления, в атмосфере аргона при нагревании по следующему режиму: нагрев до 523 К, выдержка 0,5 ч, нагревание до 873 К, выдержка 1 ч, остывание с печью. Далее продукты разложения подверглись рентгенофазовому и электронномикроскопическому (СЭМ, JEOL JSM-7001F) анализу. Рентгенограммы показали, что образцы состоят из кристаллического оксида олова рутильной структуры, другие фазы в регистрируемом количестве отсутствуют.
На рис. 14 представлены снимки порошка оксида, полученного из сульфата. Порошок при небольшом увеличении имеет вид частицы осколочной, нерегулярной формы, трещиноватые, в широком диапазоне дисперсности от единиц до десятков микрометров (рис. 14а). Большее увеличение показывает, что эти частицы представляют собой комбинацию плотных, малопористых кристаллов и высокопористых агломератов из частиц и сростков размером около 40-60 нм и развитой удельной поверхностью. Последние могут быть в виде отдельных частиц, но в основном образуют рыхлое покрытие поверхности крупнокристаллических частиц (рис. 14 б). Очевидно, что для синтеза порошка SnO2 из сульфата олова с повышенными характеристиками дисперсности и узкого распределения частиц по размерам следует подбирать режимы процесса, применять стабилизаторы и пр.
Заключение
На основании изложенного можно заключить, что наиболее эффективными являются способы получения порошков диоксида олова термолизом осадков соединений, образующихся в результате осуществления реакций в жидкой фазе. В данном случае практически нет предпочтений относительно исходных реагентов. Как показывают экспериментальные данные, изменение параметров эксперимента полностью определяет свойства целевого продукта. Стрем-– 208 – ление к высокой дисперсности конечных порошковых форм обусловливает применение ПАВ в случае как неорганических, так и органических прекурсоров. Кроме того, допирование примесями также может быть рассмотрено как один из путей блокирования роста частиц оксидной фазы. Большое значение имеет температура синтеза продуктов при термическом разложении. Следует учитывать также в каждом конкретном применении порошков SnO2, полученных из хлоридов, присутствие существенных количеств хлора даже в тщательно отмытом и отожженном ксерогеле.
В отношении процессов термического разложения рассмотренных систем можно заключить, что, несмотря на имеющиеся исследования современного уровня, очевидна недостаточная изученность этого вопроса даже в отношении многих широко используемых термически нестабильных соединений. Отсутствие надежных и однозначных литературных данных исключает возможность достаточно обоснованных априорных предсказаний и требует экспериментальных проверок в отношении каждого конкретного процесса и условий.
Авторы благодарят С.Д. Кирика и Г.М. Зеер за проведение рентгенофазового и электронномикроскопического анализа.
Статья выполнена в рамках проекта 2.1.2/531 АВЦП «Развитие научного потенциала высшей школы (2009-2010 годы)» и опубликована при поддержке Программы развития Сибирского федерального университета.
