Поражение сердца при прогрессирующей мышечной дистрофии Дюшенна (клиническое наблюдение)
")
Автор: Маянская С.Д., Булашова О.В., Мухитова Э.И., Хазова Е.В.
Журнал: Евразийский кардиологический журнал @eurasian-cardiology-journal
Рубрика: Клинический случай
Статья в выпуске: 1, 2026 года.
Бесплатный доступ
Прогрессирующая мышечная дистрофия Дюшенна – это распространённая Х-сцепленная рецессивная миопатия, детерминированная мутацией гена, кодирующего дистрофин. Миодистрофия Дюшенна сопровождается поражением сердечной мышцы с нарушением проводимости и ритма сердца, развитием дилатационной кардиомиопатии, которая может протекать бессимптомно. Развитие сердечной недостаточности определяет неблагоприятный прогноз пациентов с дистрофией Дюшенна. Недостаточная осведомлённость врачей об особенностях поражения сердца при дистрофии Дюшенна создаёт предпосылки для описания интересных клинических наблюдений. В статье представлено клиническое наблюдение развития дилатационной кардиомиопатии и сердечной недостаточности с низкой фракцией выброса левого желудочка у пациента мужского пола 19 лет с миодистрофией Дюшенна. Описывается динамика прогрессирования миодистрофии Дюшенна с момента постановки диагноза до развития декомпенсации сердечной недостаточности. Представлены данные клинического течения, лабораторные параметры, данные электрокардиографии и эхокардиографии в динамике. Приведены диагностические критерии дистрофии Дюшенна, перспективы терапии, направленной на исправление генетического дефекта путём замены, модификации или репарации гена дистрофина. Представленный случай демонстрирует необходимость персонифицированного мультидисциплинар ного подхода к пациенту с миодистрофией Дюшенна, с участием хирурга-ортопеда, пульмонолога, кардиолога, эндокринолога, офтальмолога.
Прогрессирующая мышечная дистрофия Дюшенна, дилатационная кардиомиопатия, сердечная недостаточность, ЭКГ признаки, клинический случай
Короткий адрес: https://sciup.org/143185404
IDR: 143185404 | УДК: 616.12+616-056.7 | DOI: 10.38109/2225-1685-2026-1-64-69
Heart failure in Duchenne's progressive muscular dystrophy (case report)
Progressive Duchenne muscular dystrophy is a common X-linked recessive myopathy caused by a mutations in the gene encoding dystrophin. Duchenne muscular dystrophy is characterized by damage to the heart muscle with conduction and rhythm disturbances, and the development of dilated cardiomyopathy, which may be asymptomatic. The development of heart failure determines the unfavorable prognosis of patients with Duchenne muscular dystrophy. Insufficient awareness of physicians about the characteristics of heart damage in Duchenne muscular dystrophy creates the prerequisites for describing interesting clinical observations. The article presents a clinical observation of the development of dilated cardiomyopathy and heart failure with a low left ventricular ejection fraction in a 19-year-old male patient with Duchenne muscular dystrophy. The dynamics of Duchenne muscular dystrophy progression from the moment of diagnosis to the development of decompensated heart failure are described. The article presents clinical course data, laboratory parameters, electrocardiography and echocardiography data in dynamics. The article presents diagnostic criteria for Duchenne muscular dystrophy, prospects for therapy aimed at correcting the genetic defect by replacing, modifying or reparating the dystrophin gene. The presented case demonstrates the need for a personalized multidisciplinary approach to a patient with Duchenne muscular dystrophy, with the participation of an orthopedic surgeon, pulmonologist, cardiologist, endocrinologist, and ophthalmologist.
Текст научной статьи Поражение сердца при прогрессирующей мышечной дистрофии Дюшенна (клиническое наблюдение)
Данная статья распространяется на условиях «открытого доступа», в соответствии с лицензией CC BY-NC-SA 4.0 («Attribution-NonCommercial-ShareAlike» / «Атрибуция-Не-коммерчески-Сохранение Условий» 4.0), которая разрешает неограниченное некоммерческое использование, распространение и воспроизведение на любом носителе при условии указания автора и источника. Чтобы ознакомиться с полными условиями данной лицензии на русском языке, посетите сайт: by-nc-sa/4.0/
This is an open access article distributed under the terms of the Creative Commons Attribution-NonCommercial-ShareAlike 4.0 International (CC BY-NC-SA 4.0) License , which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Миодистрофия Дюшенна (МД) относится к орфанным прогрессирующим наследственным заболеваниям мышечной системы (в среднем, 13–33 случая на 100 000 родившихся) с рецессивным типом наследования, сцепленным с Х-хромосомой [1]. Мутация гена DMD приводит к дефициту белка дистрофина, отвечающего за соединение цитоскелета мышечного волокна с базальной пластинкой (внеклеточного матрикса) через белковый комплекс, что вызывает прогрессирующую гибель миофибрилл и замещение их жировой или соединительной тканью, приводя к псевдогипертрофии [1]. Одним из диагностических критериев МД является раннее значительное (в 50-100 раз) повышение уровня креатинфосфокиназы (КФК) к третьему году жизни. Биопсия мышцы выявляет патологические вариации калибра мышечных волокон, изолированные или групповые некрозы волокон с макрофагами, элементами дегенерации, часто с центральными ядрами, с нарастанием объёма жировой и соединительной ткани с прогрессированием заболевания. Диагноз подтверждают при отсутствии дистрофина при иммуногистохимическом окрашивании [2].
Примечательно, что ранний этап развития ребёнка обычно проходит без видимых отклонений, до 3-летнего возраста родители могут отметить замедление развития навыков стояния и ходьбы. Верифицируется диагноз МД чаще в возрасте 4,6 года [1], после 6 лет отмечается нарастание мышечной слабости, проксимальной атрофии, ретракции ахилловых сухожилий, кифосколиоза, использование маневра Гауэрса1. Поражение мышц, зачастую симметрично [3]. Зависимость от инвалидной коляски наступает к 10 годам. Дисфункция гладкой мускулатуры пищеварительного тракта сопутствует 21%, мочевыводящих путей – 6% пациентам с 15 лет; сердечная недостаточность (СН) – 15% пациентам, достигшим 21,5 года [1]. У 60% пациентов с МД отмечается лёгкая умственная отсталость. К обязательным признакам МД относят снижение жизненной ёмкости лёгких (ЖЕЛ), обусловленной бронхообструктивным синдромом [3]. Кардиомиопатию при МД связывают с прогрессирующим ухудшением проводимости в пейсмекерной зоне миокарда и дилатацией полостей сердца, приводим к нарушениям ритма и проводимости, тяжёлой СН [3, 4]. Типичным изменением на электрокардиограмме (ЭКГ) при МД считают отношение R/S более 1 в отведении V i , узкий глубокий зубец Q в левых грудных отведениях и RSR', либо зазубренный зубец R в V i [4].
Прогноз пациентов с МД крайне неблагоприятный, с летальным исходом на 2-3-м десятилетии жизни, при своевременной вентиляционной поддержке – на 4-м. Ведущей причиной смерти являются прогрессирующая дыхательная и СН, реже жизнеугрожающие аритмии [3].
Представляем клинический пример кардиомиопатии у пациента с МД, после получения информированного согласия на обследование и разрешение публикации персональной медицинской информации в обезличенной форме. Уникальность представленного случая миодистрофии Дюшенна в поздней неамбулаторной стадии определяется агрессивностью течения кардиомиопатии, обусловленной дефицитом белка дистрофина с критическим снижением фракции выброса (ФВ) левого желудочка (ЛЖ) со сроком дожития пациента до 22 лет, что, в принципе, встречается не так уж и часто, хотя современная медицина позволяет продлить течение заболевания даже до 30 и более лет [5].
Пациент Н., 19 лет находился на лечении в ГАУЗ «РКБ МЗ РТ» с 29.12.2015 г. по 12.01.2016 г., после экстренной госпитализации в отделение кардиологии с пребыванием в отделении реанимации и интенсивной терапии в связи с развитием острой декомпенсации СН. При поступлении жаловался на выраженную одышку инспираторного характера в покое, общую слабость, кашель с трудно отделяемой мокротой, отёки стоп и голеней. Ухудшение самочувствия отметил за месяц до госпитализации после перенесённой пневмонии.
Диагноз «Прогрессирующая мышечная дистрофия Дюшенна» был выставлен пациенту Н. в 6-летнем возрасте. Мутация гена дистрофина идентифицирована в 19 экзоне (Xp21) (13.12.2006 г.). Пациент дважды в год проходит обследование, лечение, реабилитацию. Со слов матери, при эхокардиографии (ЭХОКГ) до 10 лет признаков дилатации сердца и снижения фракции выброса (ФВ) левого желудочка (ЛЖ) не выявлялось. Признаки СН стали появляться в течение последних 5 лет (рис. 1).
|
1997 |
Родился в срок, доношенным, беременность и роды без особенностей. Семейный анамнез: младший брат здоров, орфанных заболеваний в семье не отмечалось. |
|
2000 |
2,5 года: Задержка моторного развития: начал ходить в 2,5 года. Нарушения походки по типу «утки», частые падения. |
|
2003 |
6 лет: появились утомляемость, слабость в ногах, гипертрофия голеней. Постановка диагноза ПМДД, выявление мутации (Ген дистрофина, 19 экзон (Xp21)). |
|
2007 |
10 лет: ЭХОКГ без признаков кардиомиопатии. |
|
2009 |
12 лет: Перестал самостоятельно передвигаться. |
|
2011 |
14 лет: Появление признаков СН. |
|
2015 |
19 лет: госпитализация в отделение кардиологии в связи с декомпесацией ХСН. |
|
2016 |
20 лет: повторная госпитализация в отделение кардиологии в связи с декомпесацией ХСН. |
|
2019 |
22 лет: смерть. |
Рисунок 1. Графическое представление клинического случая [собственные данные]
Figure 1. Graphical representation of a clinical case [own data]
Анамнез жизни. Пациент Н. – первый ребёнок в семье, родился в срок, доношенным. Со слов матери беременность и роды протекали без особенностей, осложнений, приёма медикаментов, вредных привычек. Раннее умственное и физическое развитие было без особенностей, однако ползать и вставать он начал с некоторым отставанием, а ходить в 2,5 года. Родители начали отмечать нарушения походки по типу «утки», в развалку, частые падения, а с 6 лет заметили проблемы с быстрой ходьбой и бегом, появление повышенной утомляемости при ходьбе, слабости в ногах, гипертрофию голеней. До 2-го класса посещал школу, а 12 лет перестал самостоятельно передвигаться и продолжил обучение на дому. Семья пациента Н. полная, благополучная. Медико-генетическая экспертиза не выявила у младшего сына (2,5 лет) признаков какого-либо заболевания нервно-мышечной системы. До пациента Н. орфанных заболеваний в семье (до третьего поколения) не отмечалось.
Пациент Н., 19 лет, диагноз: Прогрессирующая мышечная дистрофия Дюшенна
При осмотре пациента Н. состояние тяжёлое, заторможен, на вопросы отвечает медленно, положение – ортопноэ (рис. 2). Лицо обращено вниз, мимика вялая, акроцианоз. Сколиоз грудного отдела позвоночника. Мышцы бедра плохо развиты, икроножные – несколько увеличены в размерах. Голени, лодыжки и ступни уменьшены в размерах и повёрнуты внутрь, симметричная отёчность голеней и стоп. Руки слабые, вялые, пальцы прижаты к друг другу в неполный кулак. Частота дыхательных движений (ЧДД) 24 в минуту, пульс 110 ударов в минуту, артериальное давление (АД) 140/100 мм рт. ст., сатурация кислорода (SpO 2 ) 97% -днём, 95% – ночью. Тоны сердца ритмичные, глухие. В лёгких дыхание жёсткое, справа не проводится ниже 5-го, слева – ослабленное, в нижних отделах выслушиваются мелко- и среднепузырчатые влажные хрипы. Живот безболезненный, передняя брюшная стенка при пальпации пастозная, печень пальпируется на 1 см ниже правой рёберной дуги, олигурия.
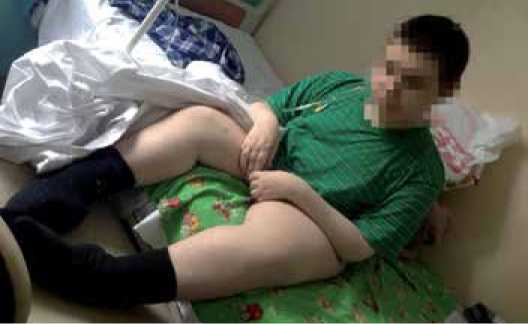
Рисунок 2. Пациент Н. Внешний вид спереди 29.12.2015 г. [собственные данные]
При ультразвуковом исследовании плевральных полостей визуализировалась гомогенная жидкость толщиной 90 мм в заднем правом синусе. На ЭКГ – синусовая тахикардия, характерный QRS комплекс в стандартных отведениях и высокие зубцы R в отведении V i ; зубцы Т уплощены в отведениях от конечностей (рис. 4).
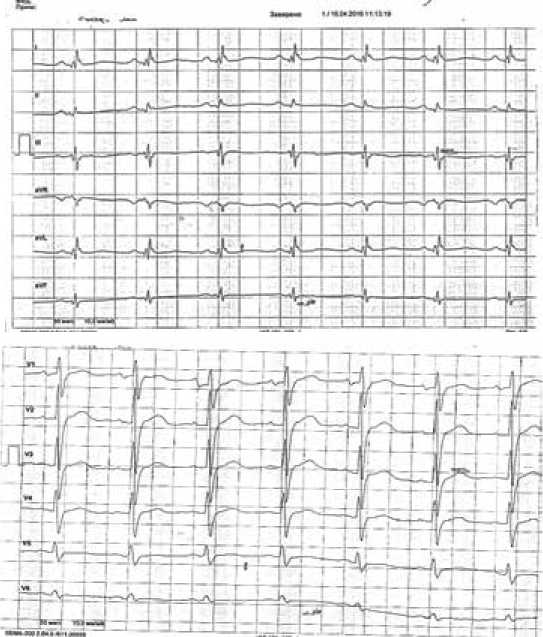
Рисунок 4. ЭКГ пациента Н. от 29.12.2015 г. [собственные данные]
Figure 4. ECG of patient N. from 29.12.2015 [own data]
Figure 2. Patient N. Frontal view, 29.12.2015 [own data]
На рентгенограмме органов грудной клетки выраженный сколиоз грудной клетки, левосторонняя полисегментарная инфильтрация, выпот в правой плевральной полости и перикарде, увеличение левых камер сердца (рис. 3).
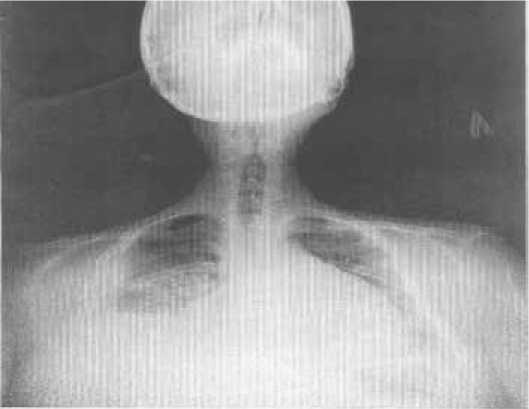
Рисунок 3. Рентгенограмма органов грудной клетки пациента Н. от 29.12.2015 г. [собственные данные]
Figure 3. Chest X-ray of patient N. from 29.12.2015[own data]
При ЭХОКГ (29.12.2015 г.): конечный диастолический размер (КДР) – 6,1 см, конечный систолический размер (КСР) – 5,5 см, толщина задней стенки ЛЖ – 0,9 см, толщина межжелудочковой перегородки – 0,8 см, правый желудочек – 3,2 см, левое предсердие – 5,1 см, ФВ ЛЖ по Симпсону – 20%, по Тейхольцу – 19%, давление в лёгочной артерии (ЛА) по трикуспидальной регургитации – 48 мм рт. ст., ширина нижней полой вены – 2,2 см, не коллабирует на вдохе. Диффузная гипокинезия миокарда ЛЖ с резким снижением его глобальной сократительной функции. Верхушечные тромбы в полости ЛЖ. Недостаточность аортального клапана 1 степени. Небольшой выпот в полости перикарда.
В общем анализе крови (30.12.2015 г.): лейкоциты – 16,4 х 109 /л (палочкоядерные нейтрофилы – 8, сегментоядерные – 62, моноциты – 10, лимфоциты – 20), СОЭ – 34 мм/ч. Общий анализ мочи (30.12.2015г.) без особенностей, протеинурии нет.
Биохимический анализ крови (29.12.2015 г.): калий – 4,0 ммоль/л, натрий – 139 ммоль/л, глюкоза – 4,0 ммоль/л, общий кальций – 2,3 ммоль/л, ионизированный кальций – 1,2 ммоль/л, фосфор – 1,38 ммоль/л, витамин D – 18 нг/мл, общий белок – 67 г/л, общий билирубин – 32,9 мкмоль/л, лактатдегидрогеназа – 330 Ед/л, гамма-глютамилтрансфераза – 143МЕ/л, креатинин – 31 мкмоль/л, мочевина – 2,8 ммоль/л, общий холестерин –
4,19 ммоль/л, аланинаминотрансфераза – 268 Ед/л, аспартатаминотрансфераза – 93Ед/л. Динамика КФК (норма 20-200 Eд/л) приведена на рисунке 5.
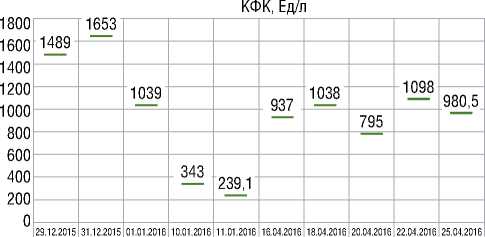
Рисунок 5. Динамика креатинфосфокиназы [собственные данные]
Figure 5. Dynamics of creatine phosphokinase [own data]
Ультразвуковое исследование органов брюшной полости выявило гепатомегалию (157 мм), почки без изменений, в надпочечниках патологические образования не визуализируются.
Таким образом, у пациента Н. с прогрессирующей МД, дилатационная кардиомиопатия с низкой ФВ ЛЖ и тромбами в области верхушки сердца осложнилась развитием острой декомпенсации хронической СН с застойной левосторонней, полисегментарной пневмонией, правосторонним гидротораксом и гидроперикардом.
Мультидисциплинарный подход при ведении пациента Н. включал консультации и согласованное мнение сосудистого хирурга (исключён тромбоз глубоких вен), невролога и пульмонолога. Проведённая терапия (антибактериальная, ингибиторы ангиотензинпревращающего фермента, бета-адреноблокаторы, диуретики, антикоагулянты, антагонисты альдостерона, глюкокортикоиды) улучшили состояние: уменьшилась одышка, жидкость в плевральной полости при выписке не визуализировалась. Рекомендовано: преднизолон 15 мг в сутки, спиронолактон 50 мг в день, фуросемид 20 мг в день, бисопролол 1,25 мг в день, варфарин 1,5 мг вечером под контролем международного нормализованного отношения 2,5-3,5, периндоприл 1,25 мг утром, ивабрадин 7,5 мг 2 раза в день.
В апреле 2016 г. пациент Н. вновь доставлен в кардиологическое отделение ГАУЗ «РКБ МЗ РТ» с жалобами на общую слабость, низкое АД, сердцебиение, одышку смешанного характера при смене положения тела, кашель с трудно отделяемой светлой, пенистой мокротой. При поступлении: положение тела – ортопноэ, ЧДД 20 в минуту, АД 90/60 мм рт. ст., пульс 90 ударов в минуту, SpO 2 - 98%, отёк лодыжек, стоп и голеней, олигурия. Тоны сердца ритмичные, дыхание ослаблено в нижних отделах, выслушиваются единичные влажные мелкопузырчатые хрипы.
По данным ЭХОКГ (16.04.2016 г.) наблюдается прогрессирование дилатации камер сердца, нарастание лёгочной гипертензии, снижение глобальной сократительной способности миокарда, увеличение предсердий правого – 5,1х4,0 см, левого –
5,4х4,3 см, расхождение листков перикарда по задней стенке 0,6 см, нижней – 1,4 см, боковой – 0,4 см, передней – 0,4 см, регургитация 2-3 степени на всех клапанах (табл. 1).
ОБСУЖДЕНИЕ
Данные литературы свидетельствуют о развитии дилатационной кардиомиопатии у 90% пациентов с МД по достижению 18-летия, при этом не всегда имеется корреляция тяжести поражения сердца со степенью слабости скелетной мускулатуры [3]. В приведённом случае признаками поражения сердца были синусовая тахикардия, характерные ЭКГ-изменения и снижение систолической функции ЛЖ, описываемые и ранее [6].
Лечение и продление жизни пациентов с МД является междисциплинарной задачей. Тактика хирурга-ортопеда направлена на мониторинг выраженности и прогрессирования сколиоза позвоночника, пульмонолога – функции внешнего дыхания для своевременного инициирования неинвазивной вентиляции. Важная роль отводится грамотному назначению преднизолона, так доза 0,75 мг/кг/сут способствует приросту мышечной массы при МД. В мультидисциплинарной команде для контроля выраженности остеопороза и функции надпочечников при постоянном использовании стероидов важная роль отводится эндокринологу, для проведения мероприятий по профилактике катаракты – офтальмологу, для оказания помощи при прогрессировании СН и аритмий – кардиологу [3].
Изучение молекулярных процессов, нарушенных из-за отсутствия дистрофина позволили получить обширные сведения о клеточной дисфункции, приводящей к прогрессирующему ухудшению состояния и преждевременной смерти пациентов. Стратегия симптоматического лечения включает ингибиторы ангиотензинпревращающего фермента, бета-адреноблокаторы, антагонисты минералокортикоидов, кортикостероиды, направленного на продление и улучшение качества жизни пациентов с МД. Инновационная (генно-таргетная) терапия обращена на изменение экспрессии функционального генного продукта, который восстановит нормальную физиологию миоцитов, т.е. терапия, потенциально излечивающая мутации, вызывающие МД, позволяя прожить полноценную жизнь [3]. Так, аталурен позволяет транслирующей рибосоме считывать информацию с мРНК, содержащей преждевременный стоп-кодон, и синтезировать полноразмерный белок. Эффективность аталурена изучена в двух рандомизированных плацебо-контролируемых исследованиях: PTC124-GD-007-DMD (ClinicalTrials.gov: NCT00592553) и PTC124-GD-020-DMD (ClinicalTrials.gov: NCT01826487) [7]. В исследовании STRIDE (307 мальчиков из 14 стран) сочетание аталурена со стандартной терапией отсрочило наступление терминальной стадии заболевания на 2,2 года (p=0,0006), потерю способности к передвижению на 4 года (p <0,0001) и снижение форсированной жизненной ёмкости лёгких до 60% и 50% на 1,8 года (p=0,0021) и 2,3 года (p=0,0207) соответственно по сравнению с применением только стандартной терапии [7]. В 2023 г. FDA2 для лечения пациентов с миодистрофией Дюшенна старше
Таблица 1. Динамика эхокардиографических параметров [собственные данные] Table 1. Dynamics of echocardiographic parameters [own data]
|
Параметр |
29.12.2015 г. |
16.04.2016 г. |
|
Конечный диастолический размер ЛЖ, см |
6,1 |
7,0 |
|
Конечный систолический размер ЛЖ, см |
5,5 |
5,8 |
|
Давление в ЛА по трикуспидальной регургитации, мм рт. ст. |
48 |
56 |
|
ФВ ЛЖ по Тейхольцу, % |
19 |
12 |
2 Food and Drug Administration, CША
68 ЕВРАЗИЙСКИЙ КАРДИОЛОГИЧЕСКИЙ ЖУРНАЛ, 1, 2026
4-х лет с подтверждённой мутацией в гене DMD, независимо от способности к передвижению, одобрило однократную рекомбинантную векторную генную терапию на основе аденоассоци-ированного вируса макак-резусов серотипа 74 (rAAVrh74) для доставки в организм гена, приводящего к выработке микроди-строфина3, который содержит выбранные домены белка дистрофина, присутствующего в нормальных мышечных клетках – де-ландистроген моксепарвовек [8].
Одна из новых стратегией терапии миодистрофии Дюшенна – редактирование/функциональное восстановление гена дистрофина с помощью CRISPR/Cas9 изучается на доклинической фазе исследования [9]. Во 2 фазе клинических испытаний лечения миодистрофии Дюшенна находится модулятор продукции утро-фина4 (Эзутромид) [10].
ЗАКЛЮЧЕНИЕ
Описанный клинический случай привлекает внимание педиатров, кардиологов к поражению сердца у пациента с прогрессирующей МД, находившемся на стандартной терапии. Ведущий вектор как в диагностике, так и в разработке новых стратегий ведения пациентов с МД для продления и улучшения качества их жизни направлен на генетические исследования.
