Поверхность потенциальной энергии системы SOCl2+2H2O

Автор: Засовская М.А., Игнатов С.К.
Журнал: Известия Коми научного центра УрО РАН @izvestia-komisc
Рубрика: Химические науки
Статья в выпуске: 2 (22), 2015 года.
Бесплатный доступ
Квантово-химическим методом рассчитаны термодинамические параметры элементарных стадий гидролиза тионилхлорида (SOCl2) в газовой фазе, протекающего с участием кластеров воды. Предполагаемый механизм включал стадии образования комплексов SOCl2 с димерами воды, в которых протекает гидролиз связи S-Cl. Показано, что участие димеров воды приводит к значительному снижению энергии активации и ускорению процесса гидролиза.
Тионилхлорид, гидролиз, теория функционала плотности, димеры воды
Короткий адрес: https://sciup.org/14992745
IDR: 14992745 | УДК: 544.18
Potential energy surface of the SOCl2+2H2O system
Thermodynamic parameters of elementary stages of thionyl chloride (SOCl2) hydrolysis in a gas phase proceeding with participation of water clusters, are calculated by a quantum-chemical method. The proposed mechanism included stages of formation of SOCl2 complexes with water dimers in which hydrolysis of S-Cl bond proceeds. Optimization of molecular geometry (search of transitive conditions) was done by a method of density functional (B3LYP//6-311++G(2d,2p)). For the found stationary points calculation of oscillatory frequencies was made and thermodynamic functions (TF) in harmonious approximation were estimated. For elementary stages of hydrolysis the transitive conditions were found and energy of activation was estimated. Results show that participation of water dimers leads to considerable decrease in energy of activation and acceleration of hydrolysis process.
Текст научной статьи Поверхность потенциальной энергии системы SOCl2+2H2O
Тионилхлорид SOCl 2 является одним из важных следовых загрязнителей атмосферы [1], гидролиз которого в атмосферном воздухе приводит к появлению сернистых и галогенсодержащих неорганических производных. Последние оказывают сильный эффект разрушения озонового слоя земли, чем объясняется интерес к изучению кинетики и механизма гидролиза SOCl 2 . Кроме того, оценка энергии связи в водных комплексах галогенидов и оксигалогенидов становится основой для совершенствования технологий очистки при получении веществ высокой чистоты, в которых продукты гидролиза являются одними из главных загрязнителей, а энергия связывания комплексов определяет теоретические пределы очистки [2].
Исследования кинетики и термодинамических характеристик гидролиза молекулы SOCl2 с участием кластеров воды немногочисленны. В экспериментальном исследовании [3] было проведено пря- мое определение порядка реакции и констант скорости гидролиза SOCl2 методом Фурье-ИК спектроскопии во влажном воздухе в газовой фазе приT=297 К. Сделан вывод, что реакция имеет первый порядок по воде и ее константа скорости составляет 6.3·10-21 см3·молекула-1·с-1. В работе [4] проведено теоретическое исследование механизма этой реакции квантовохимическими методами MP2, DFT, CCSD и QCISD. Сравнение рассчитанных констант скорости с экспериментальными значениями показало, что, несмотря на высокий уровень теории, использованной для расчета, теоретические оценки отличаются от экспериментальных результатов на несколько порядков. Для объяснения большого различия между экспериментальными и теоретическими константами скорости в работе [4] было предложено, что ускорение объясняется реакцией с димерами воды. Рассчитанные константы скорости гидролиза SOCl 2 димерами (H2O)2 в несколько раз выше, чем в случае реакции с отдельными молекулами H2O. Но полного согласия с эксперименталь- ными результатами достичь не удалось. В последующей работе [5] эта идея развита далее и были исследованы реакции гидролиза, протекающие с участием кластеров воды большего размера (H2O)п, п=1,3-7. Расчеты проведены на более высоком уровне теории (B3LYP/6-311++G(3df,3pd)), и было показано, что максимальная скорость гидролиза достигается при п=5. Однако рассчитанная константа скорости по-прежнему недооценена на 3–6 порядков. Авторы объяснили этот факт протеканием параллельных реакций, каждая из которых увеличивает скорость процесса. Тем не менее, такое объяснение представляется маловероятным, поскольку константы скорости при п=3,4,6,7 на несколько порядков ниже, чем в случае реакции, обеспечивающей максимальную скорость. При этом во всех проведенных исследованиях оставалась практически неизученной поверхность потенциальной энергии (ППЭ) для системы SOCl2+(H2O)2. Не все стационарные точки были связаны в единый путь реакции. Для ряда переходных состояний остается неясным, соответствуют ли они предполагаемым элементарным стадиям. В работе [5] сообщалось, что авторам не удалось правильно идентифицировать переходное состояние лимитирующей стадии.
В связи с этим целью настоящей работы является детальное квантовохимическое исследование ППЭ системы SOCl 2 +2H 2 O с акцентом на установление непрерывных путей реакции минимальной энергии и установление всех стадий механизма гидролиза с участием комплексов воды.
Методика исследования
Оптимизация молекулярной геометрии осуществлялась методом функционала плотности (B3LYP/6-311++G(2d,2p)). Все квантово-химические расчеты выполнены по программе Gaussian03 [6]. Для анализа результатов и термодинамических расчетов использовалась программа Moltran [7].
Результаты и обсуждения
Предполагаемый механизм гидролиза в системе SOCl 2 +2H 2 O. Долгое время считалось, что реакция гидролиза SOCl 2, соответствует простой схеме:
SOCl 2 + H 2 O → SO 2 + 2HCl. (1)
Однако теоретические оценки скорости, полученные для таких реакций квантовохимическими методами высокого уровня, отличаются от экспериментальных значений на несколько порядков. Для объяснения такого расхождения было выдвинуто предположение, что в газовой фазе в реакцию вступают не одна, а п- ное количество молекул воды, т.е. кластер (H 2 O) n :
SOCI2 + пH2O ^ SOCI2 (H2O)n
SOCl2(H2O)n→ SOCl(OH)(H2O)n-1+ HCl.(3)
Но все же и эти реакции не являются элементарными. Можно допустить, что механизм включает несколько основных элементарных стадий:
SOCl2 + H2O → SOCl2·H2O(4)
SOCl2·H2O → SOCl(OH)·HCl(5)
SOCl2·H2O + H2O → SOCl2·2H2O(6)
SOCl2 + (H2O)2→SOCl2·2H2O(7)
SOCl2·2H2O → SOCl(OH)∙H2O·HCl(8)
SOCl(OH)∙H2O·HCl → SO(OH)2∙2HCl.(9)
Образование димеров воды, участвующих в реакции (7), протекает по схеме:
2H2O → (H2O)2.(10)
Кроме того, наряду с этими реакциями, в системе SOCl 2 +2H 2 O идут процессы распада комплексов с высвобождением конечных продуктов.
Реакции (2)-(10) представляют собой предполагаемый механизм гидролиза, который рассматривается в данной работе, и эта схема является простейшим вариантом кластерного механизма. Среди этих реакций ранее фрагментарно изучались стадии (4)-(5) и (8) [4–5]. Для таких реакций были найдены переходные состояния и рассчитаны кинетические параметры. Но поверхность потенциальной энергии подробно не исследовалась, в частности, не был прослежен непрерывный путь реакции и не было доказано, что найденные стационарные точки являются единственными интермедиатами и переходными состояниями данного пути.
Дополнительно для оценки понижения энергий активации за счет взаимодействия с димерами рассмотрена стадия мономолекулярного гидролиза, соответствующая реакциям (4) и (5) с последующим образованием комплекса SOCl(OH)∙H2O·HCl за счет присоединения молекулы воды к продукту реакции (5):
SOCl(OH)∙HCl + H2O → SOCl(OH)∙H2O·HCl. (11)
Термодинамические параметры реакций гидролиза. В табл. 1 представлены термодинамические параметры предполагаемых элементарных стадий реакций гидролиза в расчете на 1 моль исходного SOCl 2 , рассчитанные на уровне B3LYP/6-311++G(2d,2p).
Из представленных данных следует, что стадии образования бинарного комплекса характеризуются типичной энергией ассоциации 15 кДж·моль-1 (энтальпия связывания 9 кДж·моль-1), присоединение второй молекулы воды с образованием тернарного комплекса более выгодно и соответствует энергии связывания 29 кДж·моль-1 (энтальпия связывания 22 кДж·моль-1). При этом энергия Гиббса остается положительной (20 кДж·моль-1 для присоединения первой молекулы воды и 17 кДж·моль-1 – для присоединения второй молекулы). Таким образом, результаты расчетов свидетельствуют, что образование тернарного комплекса несколько выгоднее образования бинарного. Это согласуется с предположением о более высокой вероятности протекания гидролиза с участием комплексов SOCl 2 · 2H 2 O.
Образование димера воды энергетически более выгодно, чем образование комплекса SOCl 2 · H 2 O, но менее выгодно, чем присоединение к нему второй молекулы воды с образованием SOCl 2 · 2H 2 O.
Анализ свободной энергии Гиббса показывает, что энергия Гиббса тримолекулярного комплекса SOCl 2 ·2H 2 O выше, чем комплекса SOCl 2 ·H 2 O, т.е. концентрация тримолекулярных комплексов в рав-
Таблица 1
Энергия, стандартные энтальпия, свободная энергия Гиббса и энтропия реакций гидролиза SOCl 2 (расчет B3LYP/6-311++G(2d,2p))
Энергия реакции
SOCl 2 ·2H 2 O → SOCl(OH)·HCl∙H 2 O на уровне B3LYP/6-311++G(2d,2p) около 14 кДж·моль-1, соответствующая энтальпия составляет 6 кДж·моль-1.
Анализ энергий гидролиза по первой и по второй связям S-Cl с образованием изолированных SOCl(OH) и H 2 SO 3 показывает, что на уровне DFT обе эти реакции слабо эндотермичны, причем гидролиз по второй связи менее выгоден. Энергия Гиббса при гидролизе по первому атому хлора меньше, чем по второму атому. Это тоже свидетельствует о том, что гидролиз по первому атому хлора более выгодный процесс по сравнению с гидролизом второй связи.
Термодинамические параметры полной реакции гидролиза (гидролиз второй связи S-Cl)
SOCl(OH)∙HCl∙H 2 O→SO(OH) 2 ∙2HCl характеризуются энергией гидролиза 28 кДж·моль-1 (энтальпия связывания 19 кДж·моль-1). Энергия Гиббса при этом также остается положительной 23 кДж·моль-1. Это подтверждает менее выгодный процесс гидролиза по второй связи.
Переходные состояния, реакционные пути и кинетические параметры гидролиза в би-и тримолекулярном комплексе. Для определения кинетических параметров был проведен поиск переходных состояний соответствующих элементарных реакций.
Мономолекулярный гидролиз в комплексе SOCl 2 ·H 2 O протекает через переходное состояние TSA, аналогичное найденным в предыдущих работах [4, 5]. Энергия активации этой реакции составляет 96.3 кДж моль-1. Продуктом этой реакции является комплекс SOCl(OH)·HCl, который может распадаться на изолированные молекулы или присоединять дополнительную молекулу H 2 O, образуя комплекс SOCl(OH)·HCl·H 2 O (обозначаемый здесь и далее как реакционный комплекс RC3). В этом комплексе происходит гидролиз по второй связи S-Cl, механизм которого одинаков для бимолекулярного (через SOCl 2 ·H 2 O) и тримолекулярного (через SOCl 2 ·2H 2 O) путей.
Поиск переходного состояния реакции гидролиза по первой связи S-Cl на поверхности потенциальной энергии системы SOCl 2 ·2H 2 O, рассчитанной на уровне B3LYP/6-311++G(2d,2p), привел к структуре TS1 (рис. 1). Построение пути реакции минимальной энергии, проводимое методом IRC [8] в направлении обратной реакции, приводит к исходному комплексу RC1. Однако неожиданно оказалось, что построение пути реакции минимальной энергии с помощью процедуры IRC в прямом направлении реакции приводит не к конечному комплексу RC3, а к новому интермедиату, обозначенному как RC2. Этот интермедиат представляет собой комплекс молекулы SOCl 2 , в которой она имеет искаженную структуру с почти плоской геометрией, координированную с H 2 O и HCl (рис.1). Длины связи S-Cl в этом комплексе хотя и заметно удлинены, остаются близки к длинам связи исходной молекулы SOCl 2 . Интермедиат RC2 распадается с образованием комплекса RC3 через еще одно переходное состояние TS2. В этом комплексе структура уже соответствует молекуле SOCl 2 с удлиненной связью S-Cl, координированной с HCl и H 2 O. Интермедиат RC2 представляет неглубокий минимум ППЭ, энергия активации реакции через TS2 составляет всего 6.2 кДж·моль-1. Таким образом, можно сделать вывод, что реакция гидролиза по первой связи протекает через последовательные стадии SOCl 2 +2H 2 O → RC1 → [TS1]≠ → RC2 → [TS2]≠ → RC3.
Подчеркнем, что для всех стадий этой схемы построены непрерывные пути реакции минимальной энергии (расчет B3LYP/6-311++G(2d,2p)), которые исключают возможность существования каких-либо дополнительных стационарных точек ППЭ этой системы.
Гидролиз по второй связи S-Cl протекает путем обычной мономолекулярной реакции
RC3 → [TS3]≠→ PC, в которой PC обозначает продуктовый комплекс SO(OH) 2 ·2HCl. На уровне B3LYP/6-311++G(2d,2p) существуют две структуры, изображенные на рис.1 как PCa и PCb, причем процедура IRC приводит из TS3 только к PCa. PCb на этом уровне теории представляет собой побочный продукт, образова-
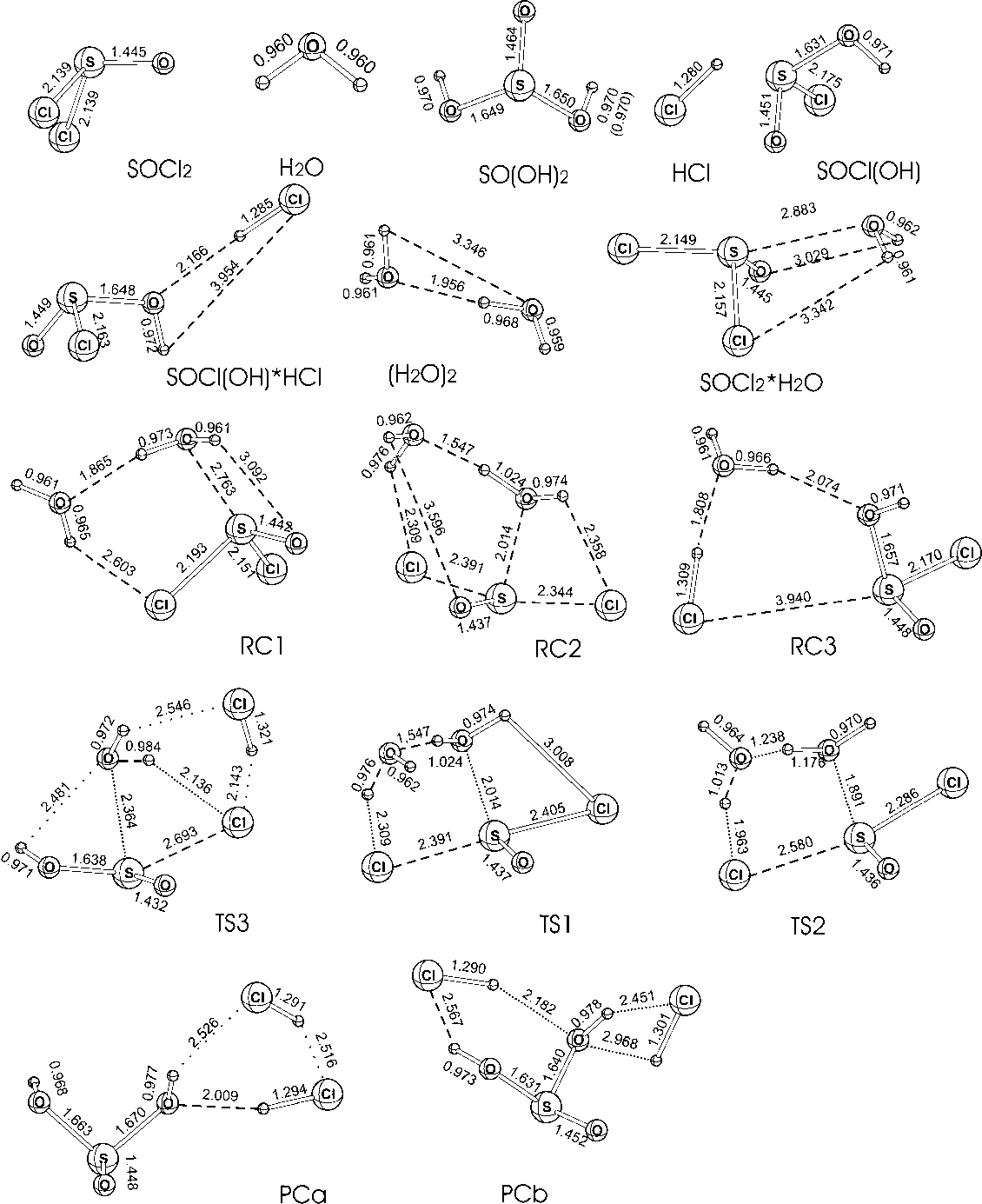
Рис. 1. Структуры реагентов (SOCl2, H2O), продуктов (SO(OH)2, HCl) и интермедиатов гидролиза в системе SOCl2+2H2O, оптимизированные на уровне B3LYP/6–311++G(2d,2p).
ние которого представляет собой параллельную реакцию (эта реакция здесь не рассматривается). Несмотря на это, и независимо от вида структуры (PCa или PCb), комплекс должен распадаться на изолированные продукты H 2 SO 3 и HCl по одному из каналов диссоциации
SO(OH) 2 ·2HCl → H 2 SO 3 ·HCl + HCl → H 2 SO 3 +2 HCl, или
SO(OH) 2 ·2HCl → H 2 SO 3 +(HCl) 2 → H 2 SO 3 +2 HCl, причем комплекс H 2 SO 3 ·HCl заметно стабильнее, чем (HCl) 2 на обоих уровнях расчета.
Структуры и геометрические параметры всех найденных стационарных точек, оптимизированные методом DFT, приведены на рис.1. Все описанные выше стадии вместе с параллельными стадиями бимолекулярного гидролиза SOCl 2 ·H 2 O изображены на диаграмме ППЭ (рис.2). Между всеми стационарными точками ППЭ, изображенными на рис. 2, рассчитаны непрерывные пути реакции с помощью процедуры IRC на уровне B3LYP/6-311++G(2d,2p)).
Как видно из диаграмм рисунков 2 и 3, бимолекулярный гидролиз по первой связи S-Cl в тернарном комплексе SOCl 2 ·2H 2 O имеет более низкий барьер активации по сравнению с бимолекулярной реакцией как с точки зрения энергии реакции, так и с точки зрения ∆ G 0(298). Причем энергия активации тримо-лекулярной реакции понижается по сравнению с бимолекулярным механизмом более чем на 50 кДж моль-1. Таким образом, при высокой температуре должна превалировать реакция в бимолекулярном комплексе, в то время как при более низких температурах выгоднее становится реакция в тримолеку-лярном комплексе. В последнем случае лимитирующей стадией становится стадия гидролиза второй связи S-Cl, протекающая через TS3. Это может приводить к различному соотношению продуктов гидролиза при высоких и низких температурах и, в принципе, может служить путем экспериментального подтверждения результатов, полученных в данной работе.
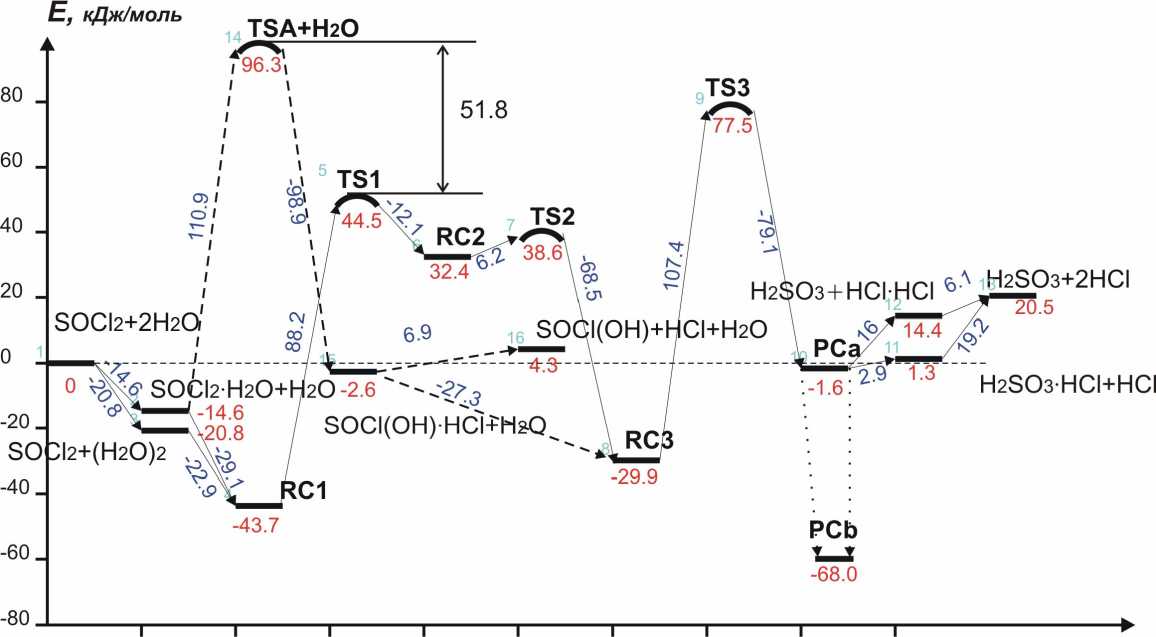
Рис.2. Диаграмма потенциальной энергии системы SOCl2+2H2O (расчет B3LYP/6–311++G(2d.2p)).
Таким образом, можно с уверенностью сказать, что рис.2 представляет собой непрерывный путь реакции (7), для которого нет дополнительных, кроме описанных выше, элементарных стадий.
Анализ диаграммы ППЭ показывает, что лимитирующей стадией реакции (7) является стадия
RC3 → TS3 → PC, энергия активации которой составляет 107.3 кДж моль-1 (энергия переходного состояния относительно исходных реагентов 77.5 кДж моль-1). Энергетические и термодинамические параметры активации всех найденных переходных состояний даны в табл. 2.
На рис.3 представлена диаграмма изменения стандартной энергии Гиббса ∆ G 0(298) для всех найденных в данной работе стационарных точек ППЭ.
Заключение
Квантовохимическим методом (B3LYP/6-311++G(2d,2p) изучены элементарные стадии газофазного гидролиза реакции тионилхлорида, протекающего в комплексах с одной и двумя молекулами воды. Определены термодинамические параметры элементарных стадий гидролиза и построены непрерывные пути реакции, соединяющие все найденные на ППЭ стационарные точки.
Полученные данные подтверждают сделанные ранее выводы о том, что механизм гидролиза в тримолекулярном комплексе имеет более низкие активационные барьеры как с точки зрения энергии, так и свободной энергии Гиббса. Энергия активации
Таблица 2
Рассчитанные (B3LYP/6–311++G(2d,2p)) полные энергии переходных состояний (Etot ≠ . а.е.), стандартные кинетические параметры элементарных стадий гидролиза SOCl 2 (кДж/моль) и мнимые частоты переходных состояний (см-1)
|
E a. кДж ∆H≠(298) кДж ∆G≠(298) кДж ∆S≠(298) Дж Реакция моль–1 моль–1 моль–1 K-1 моль–1 |
ν im см –1 |
|
RC1 →[TS1] → RC2 88.2 85.1 99.0 46.6 RC2→[TS2] → RC3 6.2 –5.6 a 0.6 –20.8 RC3→[TS3] → PC 107.3 103.8 122.9 –64.1 |
104 i 470 i 107 i |
Примечание. Отрицательное значение возникает как результат неточного учета термических поправок в гармоническом приближении для существенно ангармонической системы. При практических расчетах кинетических характеристик это значение должно быть принято за нулевое.
G, кДж/моль
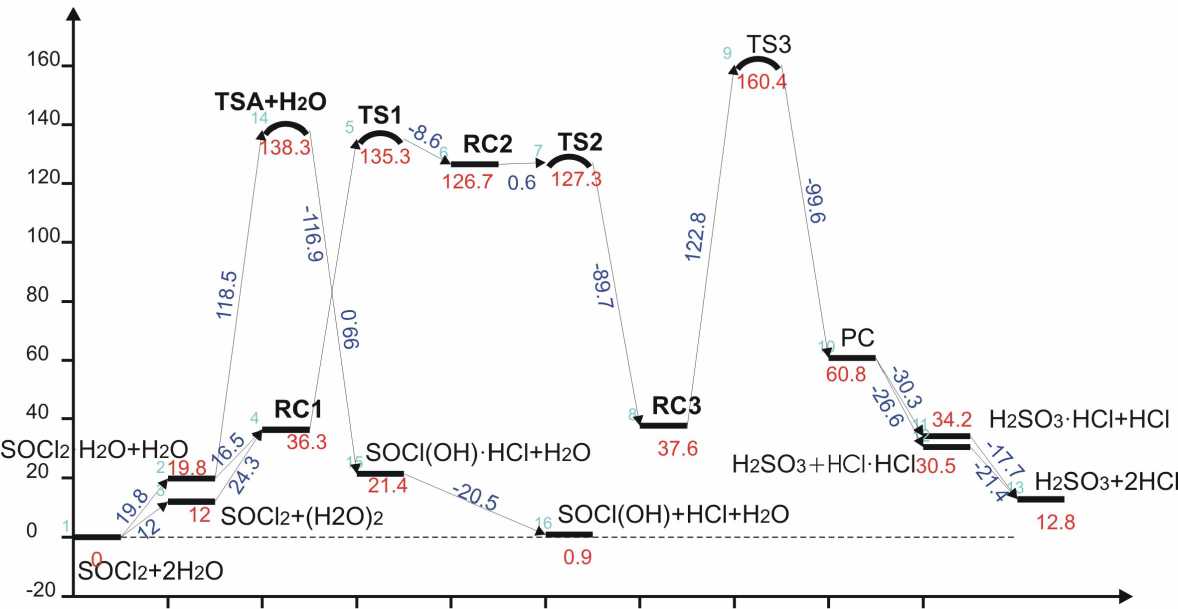
Рис.3. Диаграмма энергии Гиббса системы SOCl 2 +2H 2 O (расчет B3LYP/6–311++G(2d.2p).
в этом случае ниже энергии активации бимолекулярной реакции на 52 кДж моль-1. Лимитирующей стадией тримолекулярного гидролиза является стадия гидролиза по второй связи S-Cl. Ее энергия активации относительно SOCl 2 +2H 2 O составляет 77.5, энергия активации Гиббса –160.4 кДж моль-1.
По сравнению со стадией бимолекулярного гидролиза, тримолекулярный гидролиз должен иметь несколько иное распределение продуктов гидролиза по первой и второй связями S-Cl, что в принципе может быть зарегистрировано в эксперименте. Обнаружено, что гидролиз по первой связи S-Cl протекает через последовательность двух элементарных стадий через локальный минимум на ППЭ RC2, в котором SOCl 2 координирован с водой и имеет почти плоскую структуру.
Работа выполнена при поддержке РФФИ (проект № 14-03-00585).
Список литературы Поверхность потенциальной энергии системы SOCl2+2H2O
- Finlayson-Pitts, B.J.; Pitts, J.N., Jr. Chemistry of the Upper and Lower Atmosphere. Academic Press: New York. 1999. p. 289
- Devyatykh, G.G.; Bulanov, A D.; Gusev, A.V.; Sennikov, P.G.; Prokhorov, A.M.; Dianov, E.M.; Pohl, H.-J.: Synthesis of High-Purity Monoisotope Silicon-28//Doklady Akademii Nauk. 2001. Volume 376. № 4-6. p 46
- Johnson, T.J.; Disselkamp, R.S.; Su, Y.-F.; Fellows, R.J.; Alexander, M.L.; Driver, C.L.: Gas-phase Hydrolysis of SOCl2: Implications for Its Atmospheric Fate//J. Phys. Chem. A. 2003. №107. pp. 6183-6190
- Ignatov, S.K.; Sennikov, P.G.; Razuvaev.: Ab initio and DFT study of the molecular mechanisms of SO3 and SOCl2 rections with water in the gas phase//J. Phys. Chem. A. 2004. №108. pp. 3642-3649
- Yeung, C.S.; Ng, P.S; Guan,X.; Phillips, D.L.: Water-Assisted Dehalogenation of Thionyl Chloride in the Presence of Water Molecules//J. Phys. Chem. A. 2010. №114. pp. 4123-4130
- M.J. Frisch, G.W. Trucks, H.B. Schlegel, G.E. Scuseria, M.A. Rob, J.R. Cheeseman, J.A. Montgomery Jr., T. Vreven, K.N. Kudin, J.C. Burant, J.M. Millam, S.S. Iyengar, J. Tomasi, V. Barone, B. Mennucci, M. Cossi, G. Scal- mani, N. Rega, G. A. Petersson, H. Nakatsuji, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, M. Klene, X. Li, J.E. Knox, H.P. Hratchian, J.B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J.W. Ochterski, P.Y. Ayala, K. Morokuma, G.A. Voth, P. Salvador, J.J. Dannenberg, V.G. Zakrzewski, S. Dapprich, A.D. Daniels, M.C. Strain, O. Farkas, D.K. Malick, A.D. Rabuck, K. Raghavachari, J.B. Fores- man, J.V. Ortiz, Q. Cui, A.G. Baboul, S. Clif- ford, J. Cioslowski, B.B. Stefanov, G. Liu, A. Liashenko, P. Piskorz, I. Komaromi, R.L. Martin, D.J. Fox, T. Keith, M.A. Al-Laham, C.Y. Peng, A. Nanayakkara, M. Challacombe, P.M.W. Gill, B. Johnson, W. Chen, M.W. Wong, C. Gonzalez, and J.A. Pople, Gaussian 03 (Gaussian, Inc., Wallingford, CT, 2003)
- S.K. Ignatov, Moltran v.2.5 -Program for molecular visualization and thermodynamic calculations, University of Nizhny Novgorod, 2004, http://www.unn.ru/chem/moltran
- C. Gonzalez, H. B. Schlegel: Reaction Path Following in Mass-Weighted Internal Coordinates//J. Phys. Chem. 1990. №94. pp. 5523-5527
