Применение хромато-масс-спектрометра высокого разрешения LTQ Orbitrap для определения перфторкислот в природной воде с использованием традиционного твердофазного и металл-аффинного сорбентов: разработка и оптимизация метода

Автор: Чернова Екатерина Николаевна, Кельциева О.А., Гладилович В.Г., Русских Я.В., Суходолов Н.Г., Селютин А.А., Никифоров В.А., Жаковская З.А., Подольская Е.П.
Журнал: Научное приборостроение @nauchnoe-priborostroenie
Рубрика: Масс-спектрометрия
Статья в выпуске: 1 т.23, 2013 года.
Бесплатный доступ
Работа посвящена выбору и оптимизации метода хромато-масс-спектрометрического определения перфторкарбоновых кислот, в том числе перфтороктансульфоновой кислоты, в пробах природной воды. Показано, что использование на этапе пробоподготовки высокоспецифичного метода металл-аффинной хроматографии с применением сорбентов, содержащих ионы трехвалентного железа, позволяет повысить чувствительность анализа и избежать эффекта "переопределения". Оптимизированный метод хромато-масс-спектрометрической идентификации дает возможность определения перфторкарбоновых кислот в воде в концентрациях ниже 1 нг/л, что достаточно для практических целей.
Перфтороктансульфоновая кислота (pfos), хромато-масс-спектрометрический анализ, mrm-режим, металл-аффинные сорбенты, регулярные мультимолекулярные слои
Короткий адрес: https://sciup.org/14264840
IDR: 14264840 | УДК: 543
Application of chromato-mass-spectrometer of high resolution LTQ Orbitrap for determination of perfluorinated acids in natural water with the traditional solid-phase and metall-affine sorbents: method development and optimization
Our current research includes the description of optimization procedure of the mass-spectrometry method for detection of perfluoroalkyl acids in surface waters. The principle possibility of extracting perfluorooctansulfonate from water matrix by metal-affinity chromatography on the new sorbents containing iron (III) ions was shown. The optimized method of chromato-mass-spectrometry analysis provides low limits of detection below 1ng/L that allows the accurate quantification of perfluoroalkyl acids in water samples.
Текст научной статьи Применение хромато-масс-спектрометра высокого разрешения LTQ Orbitrap для определения перфторкислот в природной воде с использованием традиционного твердофазного и металл-аффинного сорбентов: разработка и оптимизация метода
Перфторкарбоновые кислоты (PFAs) — синтетические химические соединения, применяемые в производстве широко используемых фторполимеров. Перфторкарбоновые кислоты являются поверхностно-активными веществами, обладают высокой химической стабильностью, что делает их идеальными материалами для широкого применения (например, антипригарное покрытие для посуды, влаго- и пятностойкие покрытия для текстиля, смазки, упаковка пищевых продуктов, противопожарная пена и т. д.). Будучи крайне устойчивыми к биоразложению, перфторкарбоновые кислоты к настоящему времени обнаруживаются во многих объектах окружающей среды и живых организмах. Перфтороктановая кислота (PFOA) и перфтороктансульфонат (PFOS) являются наиболее часто детектируемыми загрязнителями этого класса [1, 2].
Исследования показывают, что PFAs очень медленно выводятся из организма человека — от 2 лет до 21 года, практически не подвергаются метаболизму и накапливаются в организме (в основном в почках и печени) [3]. Также они влияют на репродуктивную и эндокринную системы [4, 5]. Доказаны их канцерогенные свойства [6].
Перфторкарбоновые кислоты, а также перфтор-октансульфонат внесены в Список опасных ве- ществ, представляющих угрозу для Балтийского моря, разработанный странами — членами Хельсинской комиссии (ХЕЛКОМ), включающей и Российскую Федерацию [7]. Эти соединения признаны характерными загрязнителями экосистемы Балтийского моря и поэтому подлежат мониторингу и нормированию в биоте, воде и донных отложениях [8].
Работы по определению содержания перфторкарбоновых кислот в объектах окружающей среды, в частности в природной воде, ведутся во многих странах. По литературным данным, уровень концентраций в пробах природной воды составляет от десятков пг/л до сотен нг/л [3, 9]. Таким образом, анализ осложняется тем обстоятельством, что определяемые вещества являются минорными компонентами, а проба природной воды содержит значительное количество "экранирующих" веществ, мешающих определению аналита. В этом случае требуется максимальное исключение влияния матрицы на этапе пробоподготовки.
Наиболее часто применяемым в настоящее время методом пробоподготовки для определения следовых количеств перфторкарбоновых кислот является твердофазная экстракция на обращенно-фазовых сорбентах. При этом низкий уровень специфичности данного метода не всегда обеспечивает высокую степень извлечения аналита с одновременным снижением влияния матрицы. Соот- ветственно возникает необходимость разработки как аналитической процедуры, помогающей минимизировать влияние экранирующих аналиты соединений, так и условий наиболее эффективного масс-спектрометрического анализа содержания перфторкарбоновых кислот в образцах природной воды.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Используемые реактивы : ацетонитрил сорта "0" (Криохром); метанол марки LC-MS CHROMA-SOLV (Fluka); гексан сорт "1" (ОСЧ, Криохром); вода, очищенная с помощью системы Direct-Q (Millipore, электропроводность 0.056 мкСм/см при 25 ºС); ацетат аммония (LCMS grade, Fisher Scientific); тетранатриевая соль этилендиаминтет-рауксусной кислоты дигидрат (ЭДТА) (Fluka, Sigma Aldrige); соляная кислота; стеариновая кислота перекристаллизованная; трифторуксусная кислота, очищенная перегонкой; фильтрационная мембрана Владипор (МФАС-ОС-3, размер пор 0.45 мкм); SPE-картриджи Oasis HLB 60 mg (Waters).
Стандарты : перфтороктановая кислота
(PFOA), перфторнонановая кислота (PFNA), перфтордекановая кислота (PFDA), перфторундекановая кислота (PFUnDA), перфтороктансульфо-новая кислота (PFOS) и изотопно-меченная пер-фторокатансульфоновая кислота — (13С 4 PFOS) (Wellington).
Подготовка образца
Образцы природной воды отбирали в емкости темного стекла объемом 1 л. Пробу фильтровали (мембрана Владипор, 0.45 мкм), доводили рН до 3 (концентрированная соляная кислота), добавляли 0.25 г ЭДТА для связывания ионов металлов, которые могут мешать сорбции аналита. Через предварительно кондиционированные (промывали последовательно 10 мл метанола, 6 мл дистиллированной воды, 6 мл подкисленной до рН 3 воды) картриджи Oasis HLB 60 мг пропускали пробу; далее, не осушая, промывали 10 мл воды для удаления остатков ЭДТА. Сушили под вакуумом 30 мин. Элюировали последовательно 20 мл метанола, а затем 10 мл смеси метанол/ацетон (1/1). Концентрировали на роторном испарителе практически досуха, перерастворяли в 1 мл смеси метанола с 0.075 % водным раствором ацетата аммония (1/1).
Выделение перфтороктансульфоната (PFOS) из природной воды
Получение РММ (FeIII) сорбента
В специальной ванне на поверхность водной субфазы (раствор хлорида железа (III) в дистиллированной воде), наносят по каплям раствор ПАВ в подходящем неполярном легколетучем органическом растворителе (стеариновая кислота в гексане). Раствор ПАВ растекается по поверхности воды, образуя монослой, ограниченный бортами ванны и подвижными барьерами, которые могут регулировать поверхностное давление в образовавшемся монослое. Затем монослой сжимают до конденсированного состояния, после чего твердая подложка с постоянной скоростью погружается и поднимается перпендикулярно поверхности МС, и таким образом монослой переносится на твердую подложку, затем переносится в картридж. РММС в картридже хранятся в консервирующем растворе (0.01 % TFA) [10].
Проведение металл-аффинной сорбции
60 монослоев (370 дм2) РММС Fe(III) помещали в пустой картридж на фильтр. Сверху на расстоянии 2–3 см располагали второй фильтр, чтобы не допустить всплывания сорбента. Затем проводили кондиционирование колонки ацетонитрилом и промывку 2 % уксусной кислотой. Для оценки "проскока" PFOS к картриджу с РММС Fe(III) при помощи переходника последовательно присоединяли кондиционированный картридж Oasis HLB. Далее картриджи присоединяли к колбе через насадку Вюрца, которую соединяли с водоструйным насосом. Схема установки приведена на рис. 1. Образец — 50 нг PFOS, растворенной в 250 мл дистиллированной воды, с примерной скоростью 5 мл/мин поступал по капиллярной трубке на колонку с РММС Fe(III) и далее на Oasis HLB. После проведения сорбции картриджи отсоединяли
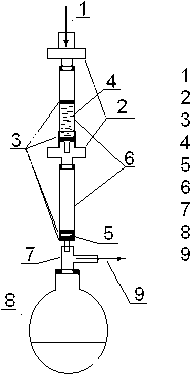
Подача образца
Переходник
Удерживающие мембраны PNIMC Ре(Ш) в водной фазе Обращенно - фазовый сорбент Картридж Насадка Вюрца Приемная колба Вакуумная .пиния
Рис. 1. Установка для извлечения PFOS из природной воды от системы, элюирование проводили раздельно. Элюирование с картриджа Oasis HLB проводили 20 мл метанола, а затем 10 мл смеси мета-нол/ацетон (1/1). Экстракт концентрировали на роторном испарителе практически досуха, пере-растворяли в 1 мл смеси 0.075 % водный раствор ацетата аммония/метанол (1/1). Перед анализом вводили аналитический стандарт — 10 нг 13С4-PFOS (10 нг), далее анализировали.
Картридж с РММС Fe(III) промывали раствором 2 % уксусной кислоты объемом 400 мкл и дистиллированной водой. Десорбцию проводили 2 % раствором уксусной кислоты в ацетонитриле (1 мл). Элюат испаряли и перерастворяли в 1 мл смеси метанола с 0.075 % водным раствором ацетата аммония (1/1), в полученный экстракт вводили аналитический стандарт 13С 4 -PFOS (10 нг), далее анализировали.
Хромато-масс-спектрометрический анализ
С использованием растворов стандартов были подобраны условия хроматографического разделения смеси аналитов на колонке С18 SupelcoSil 150 × 2.1 мм, 5 мкм. Разделение осуществляли в режиме градиентного элюирования с изменением подвижной фазы В в диапазоне от 35 % до 80 % при скорости потока 0.2 мл/мин. Состав подвижной фазы: A — 0.075 % водный раствор ацетата аммония; B — ацетонитрил, содержащий 0.075 % ацетата аммония. Масс-спектрометрический анализ проводили с использованием хромато-масс-спектрометра высокого разрешения LTQ Orbitrap фирмы Thermo Finnigan.
Масс-спектрометрический анализ выполнен в условиях электрораспылительной ионизации при регистрации отрицательных ионов, в MRM-режиме, разрешение 30 000. Фрагментацию ионов проводили в ионной ловушке в режиме столкно-вительной диссоциации (CID-режиме) при заданной варьируемой энергии столкновений в диапазоне от 5 до 35 %. Энергию фрагментации CID подбирали для получения максимальных значений интенсивностей ионов-продуктов. Для повышения точности определения масс ионов-продуктов регистрацию производили в FTMS-части (орбитальная ловушка с фурье-преобразованием). В этом случае точность определения масс не зависит от интенсивности пиков и является достаточно постоянной величиной в пределах калибровки (от недели до месяца) при неизменной температуре в помещении (при использовании внешнего стандарта — 5 ppm, при использовании внутреннего стандарта — 2 ppm).
РЕЗУЛЬТАТЫ И ОБСУЖДЕНИЕ
Современные требования к надежности качественного и количественного определения следовых количеств диктуют необходимость использования MRM — режима, при котором в узком интервале выделяется и фрагментируется ион-предшественник (родительский ион), а регистрируется только один ион-продукт (чаще всего — характеристичный сигнал, обладающий максимальной интенсивностью). В результате использования режима MRM снижается уровень шумового сигнала, повышается соотношение сигнал/шум, что приводит к значительному увеличению чувствительности анализа.
Табл. 1. Выбранная энергия фрагментации для MRM-анализа
|
Соединение |
m / z родительского иона (M–H+) |
m / z дочернего иона |
МС-МС, CID % |
Регистрируемый ион |
|
PFOA |
412.97 |
368.97604 |
25 |
[M-H+-CO 2 ]- |
|
PFNA |
462.96 |
418.97287 |
25 |
[M-H+-CO 2 ]- |
|
PFDA |
512.96 |
468.96967 |
25 |
[M-H+-CO 2 ]- |
|
PFUnDA |
562.96 |
518.96649 |
25 |
[M-H+-CO 2 ]- |
|
PFOS |
498.93 |
498.92969 |
5 * |
[M-H+]-, накопление |
|
C13-PFOS |
502.94 |
502.94169 |
5* |
[M-H+]-, накопление |
* — в связи с тем что интенсивность дочерних ионов PFOS при всех значениях CID очень мала, был выбран режим накопления молекулярного иона. При указанной энергии разбиения 5 % сигнал соэлюируемых веществ был минимален, а молекулярный ион аналита был стабилен (этой энергии было недостаточно для разбиения родительского иона аналита).
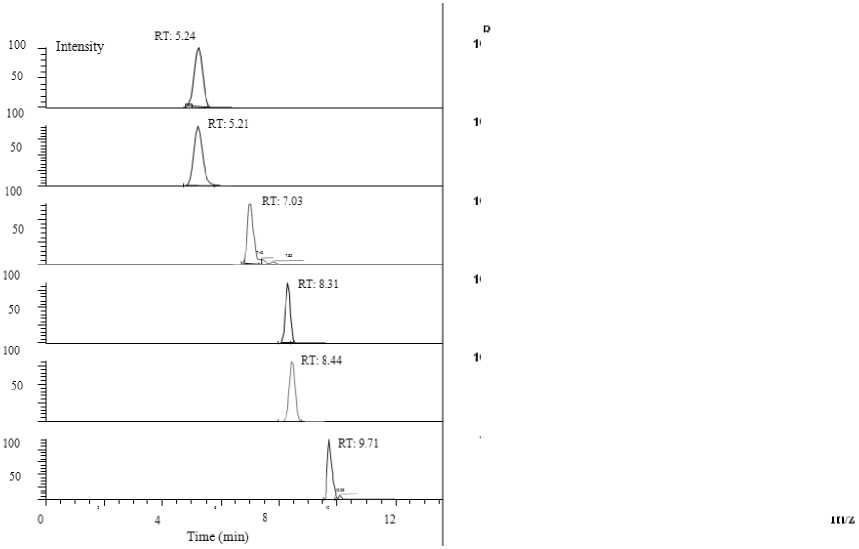
Рис. 2. Пример разделения смеси стандартов перфтрокарбоновых кислот в метаноле при оптимизированных условиях хромато-масс-спектрометрического анализа
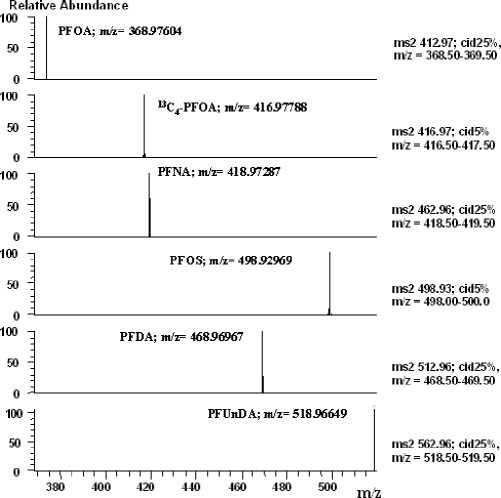
Для выбора условий эффективного хроматомасс-спектрометрического анализа была проведена оптимизация масс-спектрометрических параметров для получения максимального отклика исследуемых веществ из растворов стандартов. В режиме прямого ввода оценивали интенсивность сигнала ион-продукта каждого из аналитов. Варьировали напряжение на конусе (CV) при постоянном напряжении на ионопроводящем капилляре (ion transfer capillary) 3.2 кВ. Значительного влияния на интенсивность получаемых сигналов отмечено не было. Были выбраны следующие масс-спектрометрические параметры: напряжение на конусе (CV) — 10 В, температура ионного источника Т — 300 ºС.
Энергию разбиения CID (энергия соударений в линейной ионной ловушке) варьировали для определения максимальных значений интенсивностей дочерних ионов. Для повышения точности определения масс дочерних ионов регистрацию производили в FTMS-части (орбитальная ловушка с фурье-преобразованием). Для PFOS и ее изотопно-меченного стандарта интенсивность фрагментных ионов при всех значениях энергии фрагментации (от 5 до 45 %) оказалась слишком низкой для эффективного использования MRM-режима. Поэтому для этих соединений было принято решение использовать режим аккумулирования родительского иона при минимальной энергии разбиения
(5 %). При этих условиях наблюдалась фрагментация коэлюируемых веществ, в то время как аналит оставался в форме родительского иона. Выбранные условия фрагментации представлены в табл. 1.
Для повышения интенсивности масс-спектрометрического сигнала в режиме MRM на масс-хроматограмме выделяли сегменты, соответствующие временам выхода аналитов. В каждом сегменте сканировали ограниченное число (3–5) выбранных значений m / z ионов в диапазоне детектируемого иона ±1 а.е.м.
Mасс-хроматограмма смеси всех стандартов PFCA (0.5 нг/мл) и 13C 4 -PFOA (5 нг/мл), полученная в описанных условиях, представлена на рис. 2. Как показано на рисунке, выбранные условия позволяют достаточно эффективно разделить все выбранные аналиты.
Для проведения количественного анализа были определены концентрационные зависимости для растворов стандартов различных концентраций в диапазоне от 0.5 до 30 нг/мл в метаноле: по оси абсцисс отложена концентрация аналита, по оси ординат — отношение величин площади хроматографического пика аналита к площади хроматографического пика введенного аналитического стандарта (рис. 3). Стоит отметить, что при выбранных условиях анализа были получены линейные зависимости для всех анализируемых
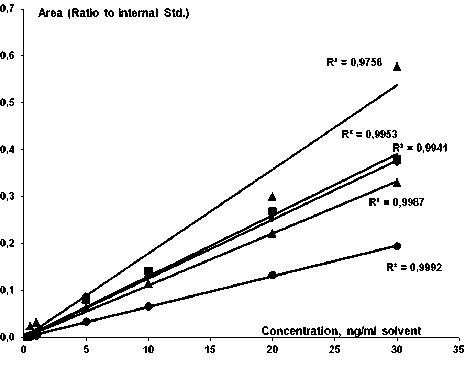
Рис. 3. Концентрационная зависимость перфтор-кислот перфторкарбоновых кислот за исключением PFOS, для которого в области низких концентраций (менее 1 нг/мл) наблюдались аномально интенсивные сигналы, не укладывающиеся в линейную зависимость. Так как целью проводимой исследовательской работы является анализ следовых количеств соединений, данная аномалия является значительной, тем не менее значение R2 = 0.97 позволяет использовать полученную калибровку для дальнейшего количественного анализа.
На этапе пробоподготовки были использованы картриджи Oasis HLB для твердофазной экстракции. Определение степени извлечения аналитов выполнено на модельной системе — раствор стандартов с концентрацией 5 нг/мл в дистиллированной воде, установленные значения приведены в табл. 2.
В связи с тем что природная матрица оказывает значительное и разнообразное влияние на степень выделения аналитов, было решено проводить параллельные исследования проб природной воды с добавкой исследуемых веществ в количестве 5 нг каждого ("spike") и исходных проб. Степень извлечения количественно оценивали методом жидкоcтной хромато-масс-спектрометрии с использованием подобранных условий анализа (табл. 2). Результаты показывают, что извлечение из природной воды происходит несколько хуже, но в достаточной степени для практического применения. Однако если для перфторкарбоновых кислот наблюдалось снижение степени экстракции, то для перфтороктаносульфоната степень извлечения составила 120 ±20 % при экстракции аналитов в концентрациях выше 1 нг. При более низких концентрациях в модельной системе ана-литов наблюдалось повышение степени извлечения до 200 %, что может быть объяснено присутствием "экранирующих", близких по массе к PFOS, соединений. На масс-хроматограмме, представленной на рис. 4, а, видно, что даже при регистрации ионов в узком диапазоне масс (2 а.е.м.) уровень фона, обусловленный содержащимися в экстракте пробы "экранирующими" соединениями, весьма высок. Соответственно для данного соединения необходимо либо подобрать отдельные параметры анализа, что весьма затруднительно, т. к. отличия между молекулярными массами аналита и коэлюируемого компонента составляют 0.02– 0.04 а.е.м., что видно на масс-спектре (рис. 4, б), либо изменить метод пробоподготовки на более специфичный.
В качестве такого метода нами была выбрана металл-аффинная хроматография, которая за все время своего развития получила применение
Табл. 2. Степень извлечения, %
|
Соединение |
Модельная система: дистиллированная вода + аналиты; pH 3, SPE |
Модельная система: природная вода + аналиты; pH 3, SPE |
|
PFOA |
54 ±16 |
44 ±7 |
|
PFNA |
87 ±10 |
45 ±10 |
|
PFDA |
80 ±14 |
50 ±10 |
|
PFUnDA |
86 ±12 |
70 ±12 |
|
PFOS |
90 ±10 |
150 ±50 |
в основном в двух областях. Первая — протеом-ный анализ, включающий в себя экстракцию, основанную на специфичном взаимодействии ме-талла—жесткой кислоты Льюиса (например, железо (III), с жестким основанием Льюиса — кислородом фосфорной группы) [11]. Вторая — анализ нуклеиновых кислот с экстракцией, основанной на связывании двухвалентной меди с атомом азота (кислота и основание Льюиса промежуточной мягкости). Стоит отметить, что наряду с кислородом к жестким основаниям Льюиса также относят и фтор. Учитывая природу PFOS, а именно наличие в молекуле как большого числа атомов фтора, так и сульфогруппы, содержащей три атома кислорода, было сделано предположение, что PFOS является объектом, который можно было бы выделить достаточно специфично из водного образца методом металл-аффинной хроматографии на сорбенте, содержащем металл—жесткую кислоту Льюиса.
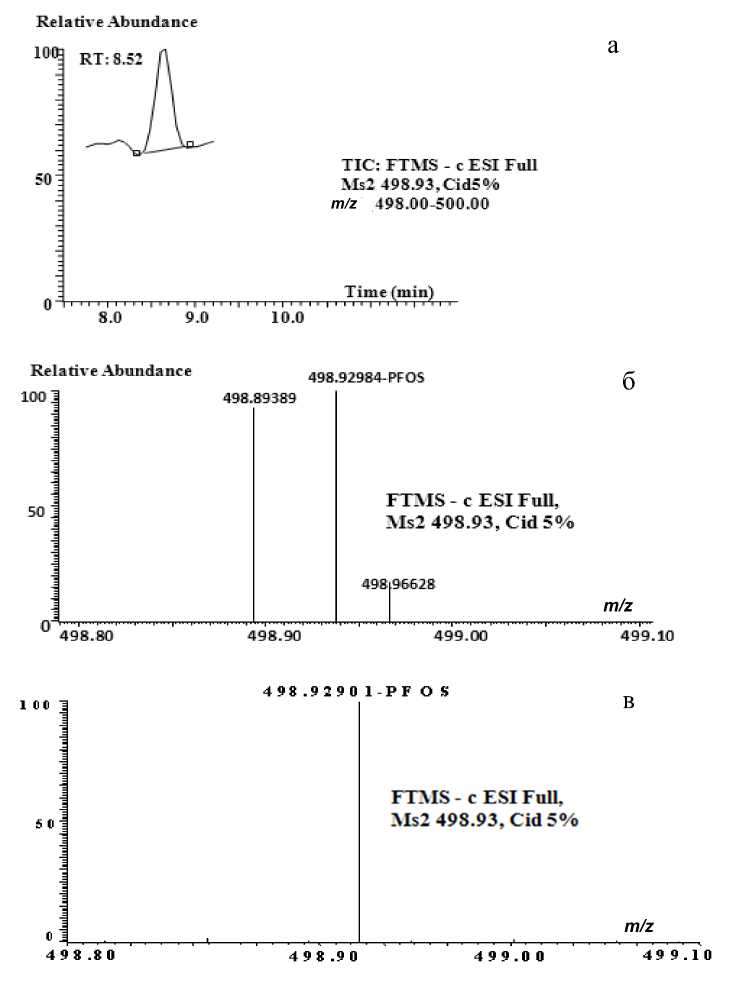
Рис. 4. Xромато-масс-спектрометрический анализ экстрактa пробы природной воды (река Нева).
а — типичная масс-хроматограмма полного ионного тока в узком диапазоне масс (2 а.е.м.); экстракция методом ТФЭ (Oasis HLB). б — масс-спектр, зафиксированный в узком диапазоне масс (2 а.е.м.); экстракция с использованием Oasis HLB. в — масс-спектр, зафиксированный в узком диапазоне масс (2 а.е.м.); экстракция с использованием РММС Fe (III)
Недавно был разработан и охарактеризован новый сорбент на основе пленок Ленгмюра— Блоджетт, содержащий ионы железа (III): регулярный мультимолекулярный сорбент на основе стеарата железа(III) — РММС Fe(III), который и был нами использован для проведения анализа [10]. Сорбент характеризуется высокой сорбционной емкостью, а также высокой специфичностью к соединениям, обладающим атомами — жесткими основаниями Льюиса. Поэтому было принято решение для исследования возможности специфичной экстракции PFOS из природной воды использовать РММС Fe(III).
Исследование проводили на следующей модельной системе: природная вода с введенным стандартом PFOS в количестве 50 нг. Экстракцию проводили с использованием установки, состоящей из картриджей с сорбентами, колбы для приема фильтрата и водоструйного насоса [7]. Скорость подачи пробы составляла 5 мл/мин. Для экстракции использовали два последовательно соединенных картриджа. Первый содержал 60 коллапсированных монослоев (450 дм2) РММС Fe(III), помещенных между двумя фильтрами таким образом, чтобы сорбент свободно находился в растворе и слои механически не взаимодействовали друг с другом. Для контроля количества не связавшейся с металл-аффинным сорбентом PFOS был использован второй картридж (Oasis HLB), т. к. предыдущий эксперимент показал хороший уровень удерживания именно PFOS на содержащемся в нем обращенно-фазовом сорбенте.
Десорбцию проводили независимо с обоих картриджей. Эксперимент показал, что с обращенно-фазового сорбента было элюировано 10 нг PFOS. В связи с этим, можно полагать, что на металл-аффинном сорбенте задержалось порядка 40 нг PFOS, т. е. величина сорбции составляет около 80 %. Анализ элюата с РММС Fe(III) показал, что содержание десорбированного аналита составляет 13 нг (степень извлечения 26 %). Вероятно, часть PFOS осталась связанной с сорбентом более прочно.
Более важным, однако, является тот факт, что в масс-спектре наблюдается единственный сигнал, соответствующий депротонированному молекулярному иону PFOS с m / z 498.93. Этот факт свидетельствует о том, что коэлюируемые компоненты в экстракте отсутствуют (рис. 4, в). Соответственно можно сделать вывод, что РММС Fe(III) действительно является сорбентом, специфичным к PFOS, и позволяет избавиться от матричных эффектов.
ЗАКЛЮЧЕНИЕ
Оптимизированная аналитическая процедура характеризуется высокой чувствительностью и селективностью. Использование MRM-режима и режима аккумулирования ионов, а также разбиение масс-хроматограммы на сегменты увеличивает чувствительность определения и снижает пределы обнаружения аналитов. Также показано, что применение на стадии пробоподготовки метода ме-талл-аффинной хроматографии на Fe(III)-содержащем сорбенте позволяет избавиться от матричных эффектов и значительно повысить селективность анализа перфторсульфоната.
Данный метод дает возможность определения перфторкарбоновых кислот в воде в концентрации менее 1 нг/л, что достаточно для большинства образцов природной воды.
