Программное моделирование прогнозирования химических свойств на основе структурной теории

Автор: Дерюгина Елена Олеговна, Чухраев Игорь Владимирович, Козеева Ольга
Статья в выпуске: 2 (12), 2018 года.
Бесплатный доступ
В статье рассмотрены существующие программные решения прогнозирования химических свойств на основе структурной теории. Выделены общие принципы функционального построения подобных систем, методы представления химических структур и расчета целевых характеристик. На основе полученных данных были выявлены причины, не позволяющие использовать существующие программы для решения задачи прогнозирования спектральных свойств соединений с учетом требований к оптимальной и эффективной организации проведения интерактивного исследования insilico.
Программы прогнозирования свойств химических соединений, органические красители, структурная теория, хромофоры
Короткий адрес: https://sciup.org/140234414
IDR: 140234414
Текст научной статьи Программное моделирование прогнозирования химических свойств на основе структурной теории
Введение. Изучение взаимосвязи структуры и функций органических красителей позволяет проводить разработку соединений с желаемыми свойствами для применения в микроскопии, для мечения белков и ДНК[1]. Органические красители широко используются в хирургической практике в диагностике и оперативном лечении для мечения тканей-мишеней [2]. Органические полупроводники (красители и пигменты, такие как метиленовый голубой, хлорофилл) обладают особенностями, которые определяются молекулярным характером их структуры, то есть присутствием сопряженных систем, которые имеют электронную или дырочнуюпроводимости, то есть обладают свойствами полупроводников, и таким образом могут быть использованы в производстве гибких и тонких пленочных фотопанелей, состоящих из двух слоев: полупроводников с положительными и с отрицательными ионами, и проводящего покрытия, пропускающего свет [3].
Достижение требуемых характеристик возможно за счет изменений в структуре соединения (хромофорного состава, связей). Для построения подобных моделей «структура-свойство» используются квантово-химические расчеты и методы машинного обучения, такие как искус-

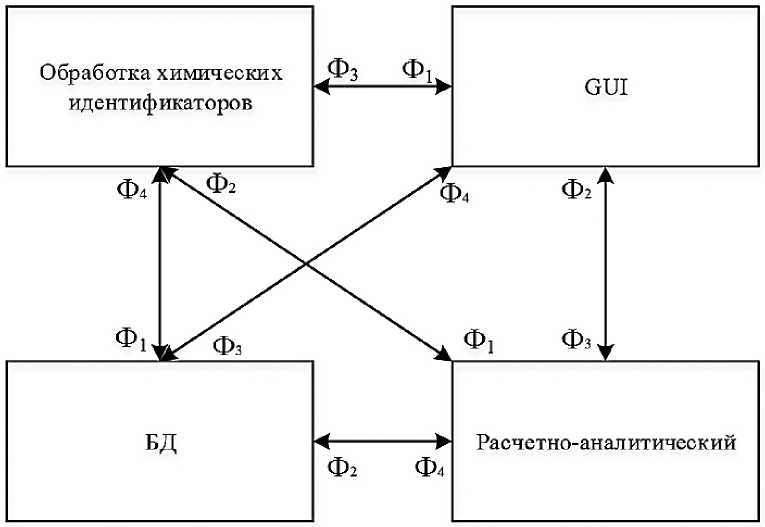
Рисунок 1. Обобщенная функциональная схема программ химического моделирования
ственные нейронные сети (ИНС), регрессионный анализ, кластеризация, деревья принятия решений. Данные методы составляют основу программных решений для прогнозирования физико-химических свойств химических соединений, которые находят свое применение при проведении научных исследований актуальных проблем химии, биологии и медицины. Целью данной статьи является изучение основных характеристик существующих программ моделирования химических свойств, применяемых в них методов реализации, а также выявления возможности использования их для прогнозирования окраски органических соединений.
Основная часть. Общими требованиями для большинства программ моделирования химических структур являются:
-
1. Работа со специфическими данными;
-
2. Наличие функций для обработки химических данных;
-
3. Наличие методов для проведения химических расчетов;
-
4. Работа с внешними базами данных;
-
5. Анализ полученных результатов;
-
6. Возможность создания интерактивной среды (пользовательского интерфейса).
Функционально подобную систему можно разделить на несколько основных модулей (Рисунок 1), описанных в Таблице 1.
Для представления информации о химическом соединении в программах используются определенные методы построения молекулярных дескрипторов, соответствующих заданному молекулярному графу. При невекторном описании применяется непосредственное кодирование матрицы смежности графа. При векторном описании молекулярного графа используются различные линейные нотации, распространенным вариантом которых является SMILES - спецификация описания состава и структуры молекулы химического вещества. SMILES представляет молекулу как последовательность символов (Рисунок 2), соответствующих определенным атомам, и заключенным в квадратные скобки (за исключением некоторых элементов), а также специальных символов, обозначающих открытие и закрытие колец и ветвей в соединении. Атомы в составе ароматических циклов обычно записываются строчными буквами вместо прописных [4].
Таблица 1.
|
Модуль |
Функции |
||
|
Обозначение |
Наименование |
Обозначение |
Описание |
|
M1 |
Обработка химических идентификаторов |
Ф1 |
Представление данных о химических соединениях в форме специальных идентификаторов |
|
M2 |
Расчетно-аналитический |
Ф2 |
Проведение расчетов, статистический анализ, прогнозирование с использованием методов машинного обучения |
|
M3 |
GUI |
Ф3 |
Визуальный ввод и вывод данных |
|
M4 |
Базы данных |
Ф4 |
Работа с локальными и внешними базами данных |
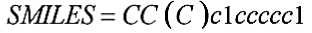

Рисунок 2. Представление структуры химического соединения в формате SMILES
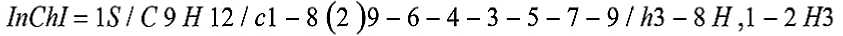
Рисунок 3. Представление структуры химического соединения в формате InChI
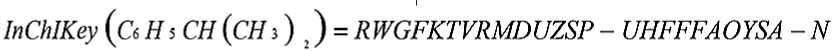
Рисунок 4 . Представление структуры химического соединения в формате InChIKey
Нотация InChI (Рисунок 3) является международным стандартом кодирования химических соединений: закодированная по данной нотации строка состоит из 6 основных «слоев» (главный, заряды, изотопный состав и другие) и нескольких «подслоев» («подслои» главного слоя: формула, перечень связанных атомов, распределение атомов водорода) [5].
Для поиска в базах данных и сети Интернет может использоваться хешированная версия InChIKey (Рисунок 4): первый набор из 14 символов кодирует строение молекулы, второй набор из 8 символов, кодирует дополнительные сведения о свойствах молекулы, флаг, обозначающий тип стандарта InChI (“S” – стандартный и “N” – нестандартный), флаг версии идентификатора (“A” - для первой), флаг, определяющий количество присоединяемых протонов [5].
Таким образом, для представления информации о структуре химического соединения используются различные методы, включающие в себя ряд идентификаторов.
Многие программы прогнозирования свойств химических соединений построены на основе ИНС; в них проводится обучение нейронного устройства, при котором определяются такие веса синапсов и порогов активации нейронов, чтобы на выходе сети формировались сигналы, соответствующие прогнозируемым значениям свойств. На вход сети подаются сигналы, описывающие химическую структуру соединения в бинарном формате MolecularACCessSystem (MACCS) chemicalfingerprint, который генерируется из начального представления химического соединения с помощью определенного идентификатора (SMILES/SMARTS, InChI). При этом используются различные архитектуры нейронных сетей, в том числе специальные, позволяющие работать, например, непосредственно с матрицами смежности молекулярных графов при невекторном описании структуры вещества [6].
В настоящее время существует ряд программ моделирования свойств химических соединений: VEGA, CORAL, ChemBench, OCHEM, Chemicalize, Ghemical и другие. Ниже кратко рассмотрены некоторые из них.
Chembench является широко используемым вебсервисом для изучения, визуализации, анализа и моделирования в области химии. Для создания моделей используются различные алгоритмы машинного обучения: «RandomForest», метод k-ближайших соседей, генетический алгоритм, алгоритм отжига. Ввод данных о химическом соединений возможен в любом формате [7].
Ochem – вычислительный сервис, работа которого основывается на таких алгоритмах машинного обучения как нейронные сети ассоциативной памяти, метод частичных наименьших квадратов, метод опорных векторов, линейный дискриминантный анализ, метод k-ближайших соседей. Информация о молекулярной структуре может быть представлена в формате SMILES, SDF или MOL2, либо найдена в соответствии с названием или идентификатором вещества в базе данных PubChem[8].
Ghemical - это программный пакет вычислительной химии, имеющий графический интерфейс пользователя и поддерживающий создание квантово-механических и молекулярных моделей с большим набором инструментов визуализации. Работа Ghemical основана на квантово-механических вычислениях с применением полуэмпириче-ских методов квантовой химии; другие функциональные возможности, такие как типизация атомов, импорт и экспорт химических файловых форматов, предоставляются с помощью системы OpenBabel [9].
Рассмотренные программные решения для прогнозирования свойств химических соединений на основе анализа их структуры в целом имеют сходные механизмы построения моделей, реализуемые с помощью методов машинного обучения и квантовой химии. Однако, при анализе существующих решений были выделены основные проблемы, не позволяющие использовать их для решения конкретной задачи прогнозирования спектральных свойств органических соединений наиболее оптимальным и визуально доступным способом с минимальными вычислительными затратами (Таблица 2).
Таблица 2.
|
Недостатки |
Возможное решение |
|
Неэффективная организация процедуры построения молекулярных структур |
Разработка среды для интерактивного формирования структур |
|
Отсутствие единого источника данных о функциональных подгруппах, обуславливающих цвет соединений |
Аккумулирование необходимых данных в единую БД |
|
Необходимость применения различных химических идентификаторов в зависимости от системы |
Конвертация всех входных идентификаторов в определенный формат |
Основным из них является отсутствие единого источника информации о функциональных подгруппах, обуславливающих окраску: данные разрознены или не об-
ладают достаточной полнотой для расчета спектральных характеристик моделируемых соединений.
В целом та или иная система имеет хотя бы один недостаток, не позволяющий использовать ее в качестве инструмента прогнозирования окраски. Одним из возможных решений данной задачи является разработка программы, в которой используется несколько методов прогнозирования, а именно прямые расчеты распределения электронного заряда в зависимости от присоединяемых хромофорных подгрупп, структура которых идентифицируется программой с помощью специального модуля, расчетов по эмпирическим правилам Физера-Вудворда и статистического анализа[10]. Кроме того, для каждого типа соединения может быть определен наиболее оптимальный метод расчета в соответствии с результатами уже проведенных вычислений на обучающей выборке и с уже описанными в литературе рекомендациями. Перечисленные методы более просты по сравнению с кванто- во-химическими расчетами, применяемыми в аналогах, но благодаря их комбинированному применению возможно получить ранжированный результат окраски, динамически отображаемый в процессе построения модели структуры соединения с минимальными требованиями к вычислительным возможностям ЭВМ.
Вывод. По результатам изучения существующих программных решений прогнозирования свойств химических соединений на основе их структуры были выделены общие принципы их реализации, а также выявлено отсутствие оптимального решения задачи моделирования окраски веществ. Поэтому актуальной является разработка программы, предназначенной в первую очередь для получения первичных результатов с видимой погрешностью прогнозирования, позволяющей проводить его в интерактивном режиме с наименьшими временными и вычислительными затратами. В перспективе, такие характеристики делают программу доступной для широкого использования.
Список литературы Программное моделирование прогнозирования химических свойств на основе структурной теории
- Hanczyc P. Applications of Chromophores and Multiphoton Techniques to Study Structure and Interactions of Bio-macromolecules in Assembled State. – Chalmers University of Technology, 2013.
- Azzopardi E. A. Chromophores in operative surgery: Current practice and rationalized development //Journal of Controlled Release. – 2017. – Т. 249. – С. 123-130
- Ashford D. L. Molecular chromophore–catalyst assemblies for solar fuel applications //Chemical reviews. – 2015. – Т. 115. – №. 23. – С. 13006-13049.
- David Weininger. SMILES, a chemical language and information system. 1. Introduction to methodology and encoding rules // J. Chem. Inf. Comput. Sci.. — 1988. — Т. 28, № 1. — С. 31-36.
- Heller S, McNaught A., Pletnev I., Stein S., Tchekhovskoi D. InChI, the IUPAC International Chemical Identifier. Journal of Cheminformatics, 2015. doi:10.1186/s13321-015-0068-4
- Myint K. Z. et al. Molecular fingerprint-based artificial neural networks QSAR for ligand biological activity predictions //Molecular pharmaceutics. – 2012. – Т. 9. – №. 10. – С. 2912-2923.
- Walker T. et al. Chembench: a cheminformatics workbench //Bioinformatics. – 2010. – Т. 26. – №. 23. – С. 3000-3001.
- Sushko I. Online chemical modeling environment (OCHEM): web platform for data storage, model development and publishing of chemical in-formation //Journal of Computer-Aided Molecular Design. – 2011. – Т. 25. – №.–С. 533-554.
- The Ghemical homepage. URL: http://bioinformatics.org/ghemical/ ghemical (датаобращения 20.11.2018)
- Козеева О.О., Чухраев И.В., Родионов А.В. Разработка на языке Python модуля поиска подструктур в химических соединениях. Электромагнитные волны и электронные системы. 2018. Т. 23. № 3. С. 57-61
